
Genome-Wide Identification and Characterization of the Oat (Avena sativa L.) WRKY Transcription Factor Family



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Genome-Wide Identification of the WRKY Gene in Oat
2.2. WRKY Classification, Phylogenetic Construction, Conserved Motifs, and Promoter Analysis of AsWRKY
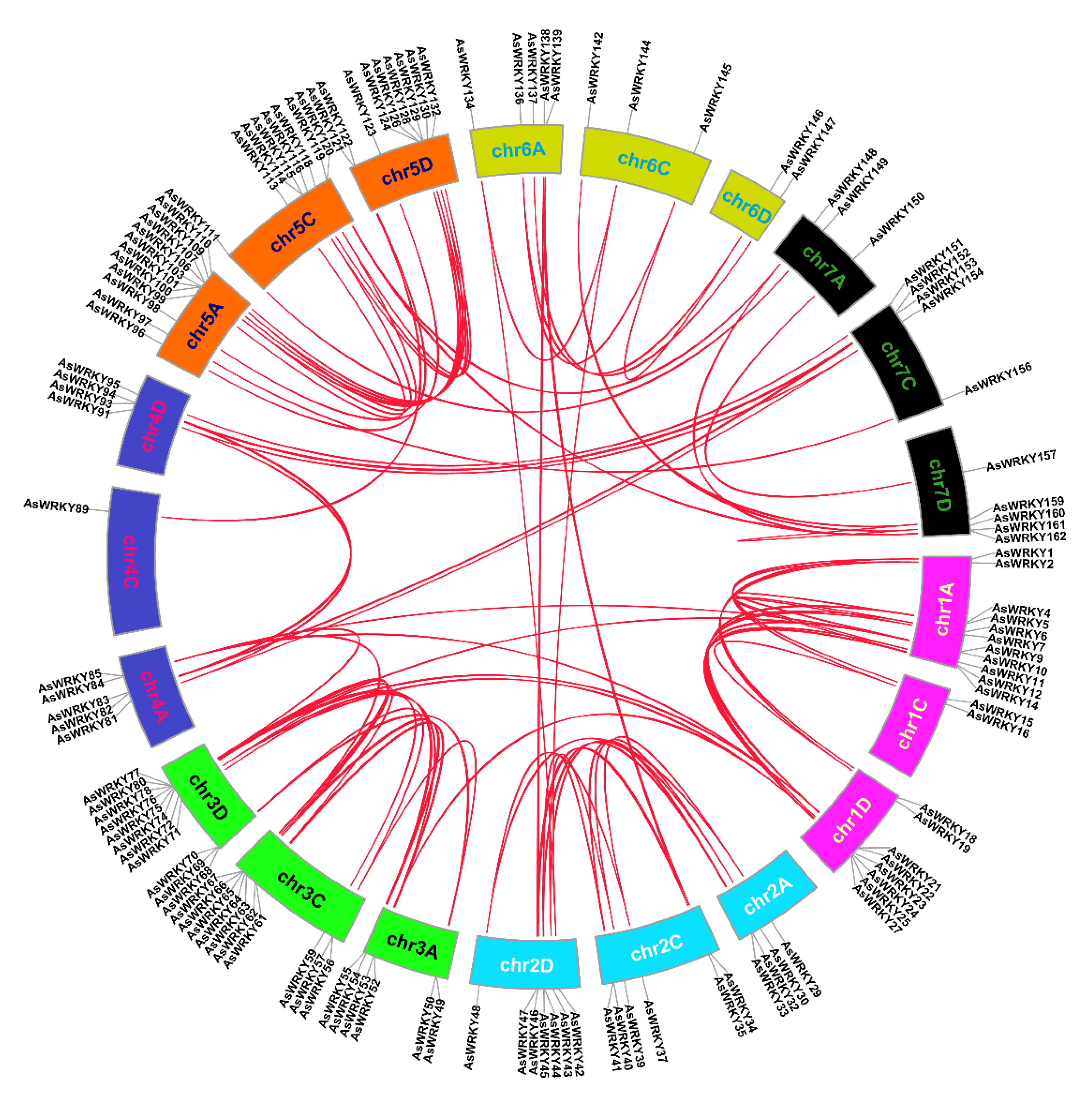
2.3. Chromosome Location and Collinearity Analysis of AsWRKY Genes
2.4. Gene Ontology and miRNA-AsWRKY Prediction
2.5. Transcriptome Expression Analysis of AsWRKY under Various Developmental Stages or Conditions
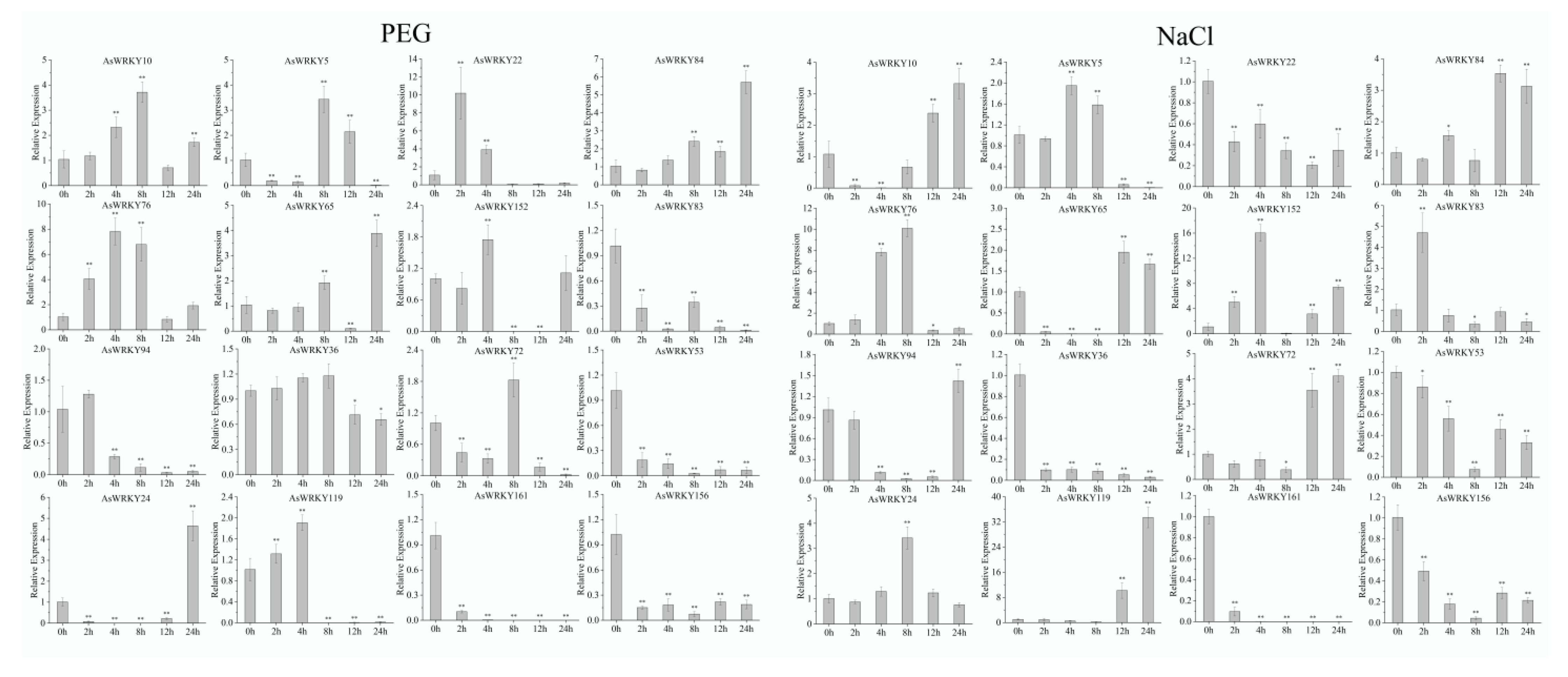
2.6. Plant Material, Growing Conditions, Drought and Salt Treatments
2.7. RNA Isolation and Expression Analysis
3. Results
3.1. Identification of the WRKY Family in Oat
3.2. Multiple Sequence Alignment, Phylogenetic Analysis, and Classification of AsWRKY Proteins
3.3. Gene Structure and Conserved Motifs Analysis of AsWRKY Genes
3.4. Chromosomal Location, Duplication, and Synteny Analysis of AsWRKY Genes
3.5. Structural Analysis of Gene Promoters
3.6. Gene Ontology of AsWRKY Proteins
3.7. miRNA Prediction of AsWRKY Interaction
3.8. Expression Profiling of Oat WRKY Genes by RNA-Seq
3.9. Expression Pattern of AsWRKY Genes in Abiotic Stress
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kilian, J.; Peschke, F.; Berendzen, K.W.; Harter, K.; Wanke, D. Prerequisites, performance and profits of transcriptional profiling the abiotic stress response. BBA Gene Regul. Mech. 2012, 1819, 166–175. [Google Scholar] [CrossRef]
- Haworth, M.; Marino, G.; Loreto, F.; Centritto, M. Integrating stomatal physiology and morphology: Evolution of stomatal control and development of future crops. Oecologia 2021, 197, 867–883. [Google Scholar] [CrossRef] [PubMed]
- Hagolani, P.F.; Zimm, R.; Vroomans, R.; Salazar-Ciudad, I. On the evolution and development of morphological complexity: A view from gene regulatory networks. PLoS Comput. Biol. 2021, 17, e1008570. [Google Scholar]
- Gahlaut, V.; Jaiswal, V.; Kumar, A.; Gupta, P.K. Transcription factors involved in drought tolerance and their possible role in developing drought tolerant cultivars with emphasis on wheat (Triticum aestivum L.). Theor. Appl. Genet. 2016, 129, 2019–2042. [Google Scholar] [CrossRef]
- Wani, S.H.; Tripathi, P.; Zaid, A.; Challa, G.S.; Kumar, A.; Kumar, V.; Upadhyay, J.; Joshi, R.; Bhatt, M. Transcriptional regulation of osmotic stress tolerance in wheat (Triticum aestivum L.). Plant Mol. Biol. 2018, 97, 469–487. [Google Scholar] [CrossRef]
- Ishiguro, S.; Nakamura, K. Characterization of a cDNA encoding a novel DNA-binding protein, SPF1, that recognizes SP8 sequences in the 5′ upstream regions of genes coding for sporamin and β-amylase from sweet potato. Mol. Genet. Genom. 1994, 244, 563–571. [Google Scholar] [CrossRef] [PubMed]
- Rushton, P.J.; Macdonald, H.; Huttly, A.K.; Lazarus, C.M.; Hooley, R. Members of a new family of DNA-binding proteins bind to a conserved cis-element in the promoters of α-Amy2 genes. Plant Mol. Biol 1995, 29, 691–702. [Google Scholar] [CrossRef] [PubMed]
- De Pater, S.; Greco, V.; Pham, K.; Memelink, J.; Kijne, J. Characterization of a zinc-dependent transcriptional activator from Arabidopsis. Nucleic Acids Res. 1996, 24, 4624–4631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Avci, U.; Nakashima, J.; Hahn, M.G.; Chen, F.; Dixon, R.A. Mutation of WRKY transcription factors initiates pith secondary wall formation and increases stem biomass in dicotyledonous plants. Plant Sci. 2010, 107, 22338–22343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imran, Q.M.; Hussain, A.; Mun, B.-G.; Lee, S.U.; Asaf, S.; Ali, M.A.; Lee, I.-J.; Yun, B.-W. Transcriptome wide identification and characterization of NO-responsive WRKY transcription factors in Arabidopsis thaliana L. Environ. Exp. Bot. 2018, 148, 128–143. [Google Scholar] [CrossRef]
- Xing, D.H.; Lai, Z.-B.; Zheng, Z.Y.; Vinod, K.; Fan, B.F.; Chen, Z.X. Stress-and pathogen-induced Arabidopsis WRKY48 is a transcriptional activator that represses plant basal defense. Mol. Plant 2008, 1, 459–470. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Zhang, L.; Yu, D. Wounding-induced WRKY8 is involved in basal defense in Arabidopsis. Mol. Plant-Microbe Interact. 2010, 23, 558–565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, L.T.; Zhong, G.M.; Wang, J.M.; Li, X.F.; Song, X.; Yang, Y. Arabidopsis WRKY28 transcription factor is required for resistance to necrotrophic pathogen, Botrytis cinerea. AFR J. Microbiol. Res. 2011, 5, 5481–5488. [Google Scholar]
- Song, Y.; Jing, S.; Yu, D. Overexpression of the stress-induced OsWRKY08 improves osmotic stress tolerance in Arabidopsis. Chin. Sci. Bul. 2009, 54, 4671–4678. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.; Shiroto, Y.; Kishitani, S.; Ito, Y.; Toriyama, K. Enhanced heat and drought tolerance in transgenic rice seedlings overexpressing OsWRKY11 under the control of HSP101 promoter. Plant Cell Rep. 2009, 28, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Jing, S.; Zhou, X.; Song, Y.; Yu, D. Heterologous expression of OsWRKY23 gene enhances pathogen defense and dark-induced leaf senescence in Arabidopsis. Plant Growth Regul. 2009, 58, 181–190. [Google Scholar] [CrossRef]
- Wai, A.H.; An, G. Histone Deacetylase 701 Enhances Abiotic Stress Resistance in Rice at the Seedling Stage by Suppressing Expression of OsWRKY45. Ph.D. Thesis, University of Manda, Yangon, Myanmar, 2018. [Google Scholar]
- Ashwini, N.; Sajeevan, R.S.; Udayakumar, M.; Nataraja, K.N.J.R.S. Identification and characterization of OsWRKY72 variant in indica genotypes. Rice Sci. 2016, 23, 297–305. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Hao, J.; Chen, X.; Hao, Z.; Wang, X.; Lou, Y.; Peng, Y.; Guo, Z. Overexpression of rice WRKY89 enhances ultraviolet B tolerance and disease resistance in rice plants. Plant Mol. Biol. 2007, 65, 799–815. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.-Q.; Yan, L.; Wu, Z.; Mei, C.; Lu, K.; Yu, Y.-T.; Liang, S.; Zhang, X.-F.; Wang, X.-F.; Zhang, D.-P. Cooperation of three WRKY-domain transcription factors WRKY18, WRKY40, and WRKY60 in repressing two ABA-responsive genes ABI4 and ABI5 in Arabidopsis. J. Exp. Bot. 2012, 63, 6371–6392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niu, C.F.; Wei, W.; Zhou, Q.Y.; Tian, A.G.; Hao, Y.J.; Zhang, W.K.; Ma, B.; Lin, Q.; Zhang, Z.B.; Zhang, J.S.; et al. Wheat WRKY genes TaWRKY2 and TaWRKY19 regulate abiotic stress tolerance in transgenic Arabidopsis plants. Plant Cell Environ. 2012, 35, 1156–1170. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Wang, R.; Zheng, M.; Liu, X.; Meng, F.; Wu, H.; Yao, Y.; Xin, M.; Peng, H.; Ni, Z.; et al. TaWRKY 51 promotes lateral root formation through negative regulation of ethylene biosynthesis in wheat (Triticum aestivum L.). Plant J. 2018, 96, 372–388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, J.; Gao, X.; Liu, Q.; Shao, Y.; Zhang, D.; Jiang, L.; Li, C. Overexpression of TaWRKY146 increases drought tolerance through inducing stomatal closure in Arabidopsis thaliana. Front. Plant Sci. 2017, 8, 2036. [Google Scholar] [CrossRef]
- Eulgem, T.; Somssich, I.E. Networks of WRKY transcription factors in defense signaling. Curr. Opin. Plant Biol. 2007, 10, 366–371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hwang, S.H.; Kwon, S.I.; Jang, J.Y.; Fang, I.L.; Lee, H.; Choi, C.; Park, S.; Ahn, I.; Bae, S.C.; Hwang, D.J. OsWRKY51, a rice transcription factor, functions as a positive regulator in defense response against Xanthomonas oryzae pv. Oryzae. Plant Cell Rep. 2016, 35, 1975–1985. [Google Scholar] [CrossRef]
- Eulgem, T.; Rushton, P.J.; Robatzek, S.; Somssich, I.E. The WRKY superfamily of plant transcription factors. Trends Plant Sci. 2000, 5, 199–206. [Google Scholar] [CrossRef]
- Rushton, P.J.; Somssich, I.E.; Ringler, P.; Shen, Q.J. WRKY transcription factors. Trends Plant Sci. 2010, 15, 247–258. [Google Scholar] [CrossRef]
- Rushton, P.J.; Torres, J.T.; Parniske, M.; Wernert, P.; Hahlbrock, K.; Somssich, I. Interaction of elicitor-induced DNA-binding proteins with elicitor response elements in the promoters of parsley PR1 genes. EMBO J. 1996, 15, 5690–5700. [Google Scholar] [CrossRef] [PubMed]
- Ülker, B.; Somssich, I.E. WRKY transcription factors: From DNA binding towards biological function. Curr. Opin. Plant Biol. 2004, 7, 491–498. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Wang, L. The WRKY transcription factor superfamily: Its origin in eukaryotes and expansion in plants. BMC Evol. Biol. 2005, 5, 1. [Google Scholar] [CrossRef] [Green Version]
- Wu, K.L.; Guo, Z.J.; Wang, H.H.; Li, J. The WRKY family of transcription factors in rice and Arabidopsis and their origins. DNA Res. 2005, 12, 9–26. [Google Scholar] [CrossRef] [Green Version]
- Machens, F.; Becker, M.; Umrath, F.; Hehl, R. Identification of a novel type of WRKY transcription factor binding site in elicitor-responsive cis-sequences from Arabidopsis thaliana. Plant Mol. Biol. 2014, 84, 371–385. [Google Scholar] [CrossRef] [PubMed]
- Ciolkowski, I.; Wanke, D.; Birkenbihl, R.P.; Somssich, I.E. Studies on DNA-binding selectivity of WRKY transcription factors lend structural clues into WRKY-domain function. Plant Mol. Biol. 2008, 68, 81–92. [Google Scholar] [CrossRef] [PubMed]
- Brand, L.H.; Fischer, N.M.; Harter, K.; Kohlbacher, O.; Wanke, D. Elucidating the evolutionary conserved DNA-binding specificities of WRKY transcription factors by molecular dynamics and in vitro binding assays. Nucleic Acids Res. 2013, 41, 9764–9778. [Google Scholar] [CrossRef] [PubMed]
- He, H.; Dong, Q.; Shao, Y.; Jiang, H.; Zhu, S.; Cheng, B.; Xiang, Y. Genome-wide survey and characterization of the WRKY gene family in Populus trichocarpa. Plant Cell Rep. 2012, 31, 1199–1217. [Google Scholar] [CrossRef] [PubMed]
- Ramamoorthy, R.; Jiang, S.Y.; Kumar, N.; Venkatesh, P.N.; Ramachandran, S. A comprehensive transcriptional profiling of the WRKY gene family in rice under various abiotic and phytohormone treatments. Plant Cell Rep. 2008, 49, 865–879. [Google Scholar] [CrossRef]
- Ning, P.; Liu, C.; Kang, J.; Lv, J. Genome-wide analysis of WRKY transcription factors in wheat (Triticum aestivum L.) and differential expression under water deficit condition. PeerJ 2017, 5, e3232. [Google Scholar] [CrossRef] [Green Version]
- Ross, C.A.; Liu, Y.; Shen, Q. The WRKY gene family in rice (Oryza sativa). J. Integr. Plant Biol. 2007, 49, 827–842. [Google Scholar] [CrossRef]
- Bencke Malato, M.; Cabreira, C.; Wiebke Strohm, B.; Bücker Neto, L.; Mancini, E.; Osorio, M.B.; Homrich, M.S.; Turchetto Zolet, A.C.; de Carvalho, M.C.; Stolf, R. Genome-wide annotation of the soybean WRKY family and functional characterization of genes involved in response to Phakopsora pachyrhiziinfection. BMC Plant Biol. 2014, 14, 236. [Google Scholar] [CrossRef] [Green Version]
- Wei, K.F.; Chen, J.; Chen, Y.F.; Wu, L.J.; Xie, D.X. Molecular phylogenetic and expression analysis of the complete WRKY transcription factor family in maize. DNA Res. 2012, 19, 153–164. [Google Scholar] [CrossRef] [Green Version]
- Gutierrez Gonzalez, J.J.; Tu, Z.J.; Garvin, D.F. Analysis and annotation of the hexaploid oat seed transcriptome. BMC Genom. 2013, 14, 471. [Google Scholar] [CrossRef] [Green Version]
- He, X.; Bjornstad, A. Diversity of North European oat analyzed by SSR, AFLP and DArT markers. Theor. Appl. Genet. 2012, 125, 57–70. [Google Scholar] [CrossRef] [PubMed]
- Han, L.; Eneji, A.E.; Steinberger, Y.; Wang, W.; Yu, S.; Liu, H.; Liu, J. Comparative biomass production of six oat varieties in a saline soil ecology. Commun. Soil Sci. Plan 2014, 45, 2552–2564. [Google Scholar] [CrossRef]
- Yao, E.; Blake, V.C.; Cooper, L.; Wight, C.P.; Michel, S.; Cagirici, H.B.; Lazo, G.R.; Birkett, C.L.; Waring, D.J.; Jannink, J.-L. GrainGenes: A data-rich repository for small grains genetics and genomics. Database 2022, 2022, baac034. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.D.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Mistry, J.; Mitchell, A.L.; Potter, S.C.; Punta, M.; Qureshi, M.; Sangrador-Vegas, A. The Pfam protein families database: Towards a more sustainable future. Nucleic Acids Res. 2016, 44, D279–D285. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; Wang, J.; Chitsaz, F.; Derbyshire, M.K.; Geer, R.C.; Gonzales, N.R.; Gwadz, M.; Hurwitz, D.I.; Marchler, G.H.; Song, J.S. CDD/SPARCLE: The conserved domain database in 2020. Nucleic Acids Res. 2020, 48, D265–D268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schultz, J.; Copley, R.R.; Doerks, T.; Ponting, C.P.; Bork, P. SMART: A web-based tool for the study of genetically mobile domains. Nucleic Acids Res. 2000, 28, 231–234. [Google Scholar] [CrossRef]
- Artimo, P.; Jonnalagedda, M.; Arnold, K.; Baratin, D.; Csardi, G.; de Castro, E.; Duvaud, S.; Flegel, V.; Fortier, A.; Gasteiger, E. ExPASy: SIB bioinformatics resource portal. Nucleic Acids Res. 2012, 40, W597–W603. [Google Scholar] [CrossRef]
- Yu, C.S.; Chen, Y.C.; Lu, C.H.; Hwang, J.K. Prediction of protein subcellular localization. Proteins Struct. Funct. Bioinform. 2006, 64, 643–651. [Google Scholar] [CrossRef]
- Horton, P.; Park, K.-J.; Obayashi, T.; Fujita, N.; Harada, H.; Adams-Collier, C.; Nakai, K. WoLF PSORT: Protein localization predictor. Nucleic Acids Res. 2007, 35, W585–W587. [Google Scholar] [CrossRef] [Green Version]
- Temnykh, S.; DeClerck, G.; Lukashova, A.; Lipovich, L.; Cartinhour, S.; McCouch, S. Computational and experimental analysis of microsatellites in rice (Oryza sativa L.): Frequency, length variation, transposon associations, and genetic marker potential. Genome Res. 2001, 11, 1441–1452. [Google Scholar] [CrossRef] [Green Version]
- Bailey, T.L.; Boden, M.; Buske, F.A.; Frith, M.; Grant, C.E.; Clementi, L.; Ren, J.; Li, W.W.; Noble, W.S. MEME SUITE: Tools for motif discovery and searching. Nucleic Acids Res. 2009, 37, W202–W208. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.; Xia, R. TBtools: An integrative toolkit developed for interactive analyses of big biological data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef] [PubMed]
- Lescot, M.; Déhais, P.; Thijs, G.; Marchal, K.; Moreau, Y.; van de Peer, Y.; Rouzé, P.; Rombauts, S. PlantCARE, a database of plant cis-acting regulatory elements and a portal to tools for in silico analysis of promoter sequences. Nucleic Acids Res. 2002, 30, 325–327. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Tang, H.; DeBarry, J.D.; Tan, X.; Li, J.; Wang, X.; Lee, T.-h.; Jin, H.; Marler, B.; Guo, H. MCScanX: A toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 2012, 40, e49. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Zhang, Y.; Zhang, Z.; Zhu, J.; Yu, J. KaKs_Calculator 2.0: A toolkit incorporating gamma-series methods and sliding window strategies. Genom Proteom Bioinf. 2010, 8, 77–80. [Google Scholar] [CrossRef] [Green Version]
- Peng, Y.; Yan, H.; Guo, L.; Deng, C.; Kang, L.; Wang, C.; Zhou, P.; Yu, K.; Dong, X.; Zhao, J. Reference genome assemblies reveal the origin and evolution of allohexaploid oat. Res. Sq. 2022, 54, 1248–1258. [Google Scholar] [CrossRef]
- Huerta-Cepas, J.; Szklarczyk, D.; Heller, D.; Hernández-Plaza, A.; Forslund, S.K.; Cook, H.; Mende, D.R.; Letunic, I.; Rattei, T.; Jensen, L.J. eggNOG 5.0: A hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res. 2019, 47, D309–D314. [Google Scholar] [CrossRef] [Green Version]
- Axtell, M.J.; Meyers, B.C. Revisiting criteria for plant microRNA annotation in the era of big data. Plant Cell 2018, 30, 272–284. [Google Scholar] [CrossRef] [Green Version]
- Bray, N.L.; Pimentel, H.; Melsted, P.; Pachter, L. Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 2016, 34, 525–527. [Google Scholar] [CrossRef]
- Cheng, H.; Ma, X.; Jia, s.; Li, M.; Mao, P. Transcriptomic analysis reveals the changes of energy production and AsA-GSH cycle in oat embryos during seed ageing. Plant Physiol. Bioch. 2020, 153, 40–52. [Google Scholar] [CrossRef]
- Wu, B.; Hu, Y.; Huo, P.; Zhang, Q.; Chen, X.; Zhang, Z. Transcriptome analysis of hexaploid hulless oat in response to salinity stress. PLoS ONE 2017, 12, e0171451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Lysøe, E.; Armarego-Marriott, T.; Erban, A.; Paruch, L.; van Eerde, A.; Bock, R.; Liu-Clarke, J. Transcriptome and metabolome analyses provide insights into root and root-released organic anion responses to phosphorus deficiency in oat. J. Exp. Bot. 2018, 69, 3759–3771. [Google Scholar]
- Zhang, J. Evolution by gene duplication: An update. Trends Ecol. Evol. 2003, 18, 292–298. [Google Scholar] [CrossRef]
- Tripathi, P.; Rabara, R.C.; Rushton, P.J. A systems biology perspective on the role of WRKY transcription factors in drought responses in plants. Planta 2014, 239, 255–266. [Google Scholar] [CrossRef] [PubMed]
- Giacomelli, J.I.; Weigel, D.; Chan, R.L.; Manavella, P.A. Role of recently evolved miRNA regulation of sunflower HaWRKY6 in response to temperature damage. New Phytol. 2012, 195, 766–773. [Google Scholar] [CrossRef] [PubMed]
- Phukan, U.J.; Jeena, G.S.; Shukla, R.K. WRKY transcription factors: Molecular regulation and stress responses in plants. Front. Plant Sci. 2016, 7, 760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lippok, B.; Birkenbihl, R.P.; Rivory, G.; Brümmer, J.; Schmelzer, E.; Logemann, E.; Somssich, I.E. Expression of AtWRKY33 encoding a pathogen-or PAMP-responsive WRKY transcription factor is regulated by a composite DNA motif containing W box elements. Mol. Plant-Microbe Interact. 2007, 20, 420–429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brand, L.H.; Kirchler, T.; Hummel, S.; Chaban, C.; Wanke, D. DPI-ELISA: A fast and versatile method to specify the binding of plant transcription factors to DNA in vitro. Plant Methods 2010, 6, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Wang, M.; Zhang, X.; Hao, B.; Kaushik, S.; Pan, Y. WRKY gene family evolution in Arabidopsis thaliana. Genetica 2011, 139, 973–983. [Google Scholar] [CrossRef]
- Giacomelli, J.I.; Ribichich, K.F.; Dezar, C.A.; Chan, R.L. Expression analyses indicate the involvement of sunflower WRKY transcription factors in stress responses, and phylogenetic reconstructions reveal the existence of a novel clade in the Asteraceae. Plant Sci. 2010, 178, 398–410. [Google Scholar] [CrossRef]
- Wei, Y.; Shi, H.; Xia, Z.; Tie, W.; Ding, Z.; Yan, Y.; Wang, W.; Hu, W.; Li, K. Genome-wide identification and expression analysis of the WRKY gene family in cassava. Front. Plant Sci. 2016, 7, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, H.; Zhu, W.; Silva, J.C.; Gu, X.; Buell, C.R. Intron gain and loss in segmentally duplicated genes in rice. Genome Biol. 2006, 7, R41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flagel, L.E.; Wendel, J.F. Gene duplication and evolutionary novelty in plants. New Phytol. 2009, 183, 557–564. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Yang, J.; Li, G.; Sun, Z.; Hu, S.; Chen, Y.; Guo, W.; Hou, H.J.G. Genome-wide identification and comparative analysis of the WRKY gene family in aquatic plants and their response to abiotic stresses in giant duckweed (Spirodela polyrhiza). Genomics 2021, 113, 1761–1777. [Google Scholar] [CrossRef]
- Lei, P.; Wei, X.; Gao, R.; Huo, F.; Nie, X.; Tong, W.; Song, W. Genome-wide identification of PYL gene family in wheat: Evolution, expression and 3D structure analysis. Genomics 2021, 113, 854–866. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Yang, E.; He, Q.; Lin, M.; Zhou, W.; Pian, R.; Chen, X. Genome-wide analysis of the WRKY gene family in drumstick (Moringa oleifera Lam.). PeerJ 2019, 7, e7063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, C.; Teixeira da Silva, J.A.; Tan, J.; Zhang, J.; Pan, X.; Li, M.; Luo, J.; Duan, J.J. A genome-wide identification of the WRKY family genes and a survey of potential WRKY target genes in Dendrobium officinale. Sci. Rep. 2017, 7, 9200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Q.; Li, Y.; Zhang, Y.; Wu, C.; Wang, S.; Hao, L.; Wang, S.; Li, T. Md-miR156ab and Md-miR395 target WRKY transcription factors to influence apple resistance to leaf spot disease. Front. Plant Sci. 2017, 8, 526. [Google Scholar] [CrossRef] [Green Version]
- Kumar, R.R.; Pathak, H.; Sharma, S.K.; Kala, Y.K.; Nirjal, M.K.; Singh, G.P.; Goswami, S.; Rai, R.D. Novel and conserved heat-responsive microRNAs in wheat (Triticum aestivum L.). Funct. Integr. Genom. 2015, 15, 323–348. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.K.; Prasad, A.; Maurya, J.; Prasad, M. Regulation of small RNA-mediated high temperature stress responses in crop plants. Plant Cell Rep. 2021, 11, 765–773. [Google Scholar] [CrossRef]
- Chen, L.; Song, Y.; Li, S.; Zhang, L.; Zou, C.; Yu, D. The role of WRKY transcription factors in plant abiotic stresses. BBA-Gene Regul. Mech. 2012, 1819, 120–128. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Fu, Q.; Chen, L.; Huang, W.; Yu, D. Arabidopsis thaliana WRKY25, WRKY26, and WRKY33 coordinate induction of plant thermotolerance. Planta 2011, 233, 1237–1252. [Google Scholar] [CrossRef] [PubMed]
- Shang, Y.; Yan, L.; Liu, Z.Q.; Cao, Z.; Mei, C.; Xin, Q.; Wu, F.Q.; Wang, X.F.; Du, S.Y.; Jiang, T. The Mg-chelatase H subunit of Arabidopsis antagonizes a group of WRKY transcription repressors to relieve ABA-responsive genes of inhibition. Plant Cell 2010, 22, 1909–1935. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, N.; Rizhsky, L.; Liang, H.; Shuman, J.; Shulaev, V.; Mittler, R. Enhanced tolerance to environmental stress in transgenic plants expressing the transcriptional coactivator multiprotein bridging factor 1c. Plant Physiol. 2005, 139, 1313–1322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, Y.; Liang, G.; Yu, D. Activated expression of WRKY57 confers drought tolerance in Arabidopsis. Mol. Plant 2012, 5, 1375–1388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, X.; Chen, Z.; Liu, Y.; Zhang, H.; Zhang, M.; Liu, Q.; Hong, X.; Zhu, J.K.; Gong, Z. ABO3, a WRKY transcription factor, mediates plant responses to abscisic acid and drought tolerance in Arabidopsis. Plant J. 2010, 63, 417–429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berri, S.; Abbruscato, P.; Faivre Rampant, O.; Brasileiro, A.; Fumasoni, I.; Satoh, K.; Kikuchi, S.; Mizzi, L.; Morandini, P.; Pè, M.E. Characterization of WRKYco-regulatory networks in rice and Arabidopsis. BMC Plant Biol. 2009, 9, 120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiu, Y.; Yu, D. Over-expression of the stress-induced OsWRKY45 enhances disease resistance and drought tolerance in Arabidopsis. Environ. Exp. Bot. 2009, 65, 35–47. [Google Scholar] [CrossRef]
- Zhou, Q.Y.; Tian, A.G.; Zou, H.F.; Xie, Z.M.; Lei, G.; Huang, J.; Wang, C.M.; Wang, H.W.; Zhang, J.S.; Chen, S.Y. Soybean WRKY-type transcription factor genes, GmWRKY13, GmWRKY21, and GmWRKY54, confer differential tolerance to abiotic stresses in transgenic Arabidopsis plants. Plant Biotechnol. J. 2008, 6, 486–503. [Google Scholar] [CrossRef]
- Wang, C.; Deng, P.; Chen, L.; Wang, X.; Ma, H.; Hu, W.; Yao, N.; Feng, Y.; Chai, R.; Yang, G. A wheat WRKY transcription factor TaWRKY10 confers tolerance to multiple abiotic stresses in transgenic tobacco. PLoS ONE 2013, 8, e65120. [Google Scholar] [CrossRef]










Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, K.; Ju, Z.; Jia, Z.; Liang, G.; Ma, X.; Liu, W. Genome-Wide Identification and Characterization of the Oat (Avena sativa L.) WRKY Transcription Factor Family. Genes 2022, 13, 1918. https://doi.org/10.3390/genes13101918
Liu K, Ju Z, Jia Z, Liang G, Ma X, Liu W. Genome-Wide Identification and Characterization of the Oat (Avena sativa L.) WRKY Transcription Factor Family. Genes. 2022; 13(10):1918. https://doi.org/10.3390/genes13101918
Chicago/Turabian StyleLiu, Kaiqiang, Zeliang Ju, Zhifeng Jia, Guoling Liang, Xiang Ma, and Wenhui Liu. 2022. "Genome-Wide Identification and Characterization of the Oat (Avena sativa L.) WRKY Transcription Factor Family" Genes 13, no. 10: 1918. https://doi.org/10.3390/genes13101918