
Episomes and Transposases—Utilities to Maintain Transgene Expression from Nonviral Vectors

Abstract
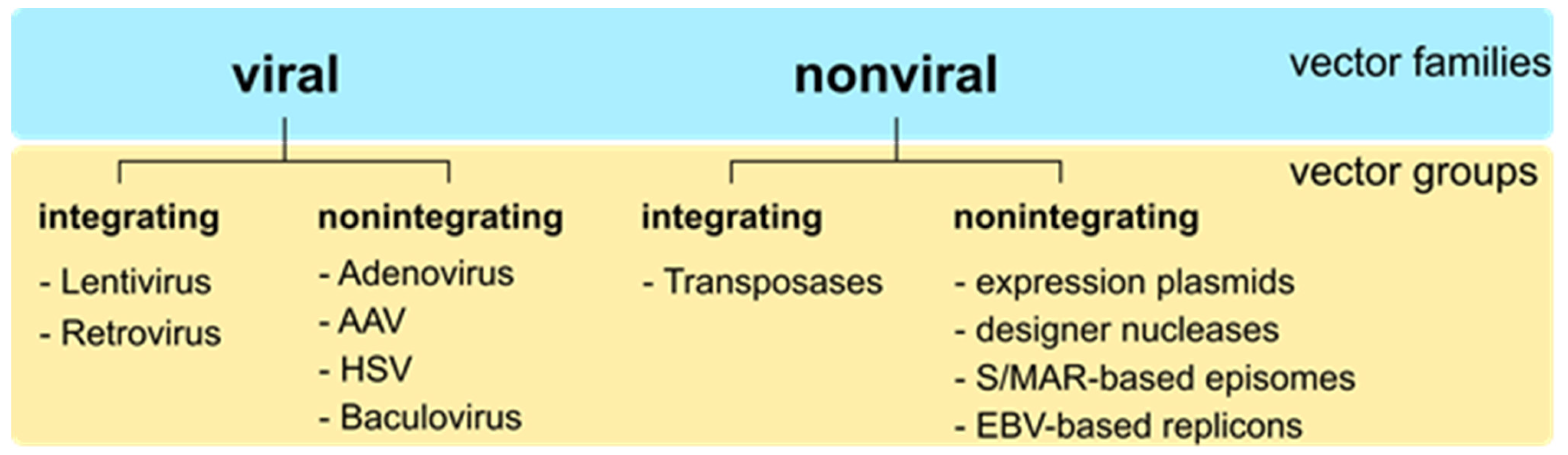
:1. Introduction
2. Transgene Expression
3. Mitotic Stability
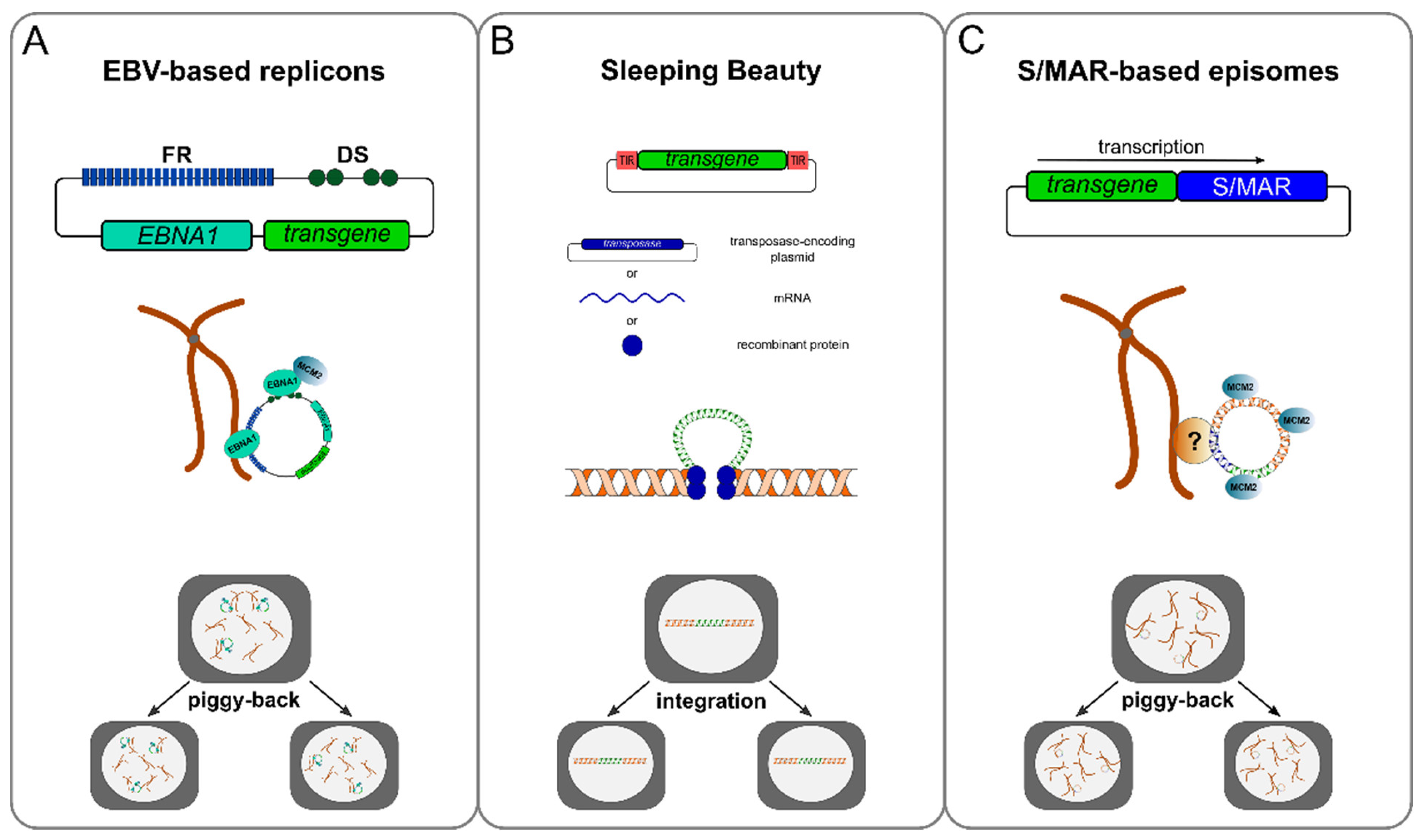
3.1. Viral Elements That Confer Mitotic Stability
3.2. Chromosomal Elements That Confer Mitotic Stability
3.3. Modifications to Minimize Effects of Innate Immune Responses
4. Nuclear Localisation
5. Nucleic Acid Delivery
6. Discussion and Outlook
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sakuma, T.; Barry, M.A.; Ikeda, Y. Lentiviral Vectors: Basic to Translational. Biochem. J. 2012, 443, 603–618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kebriaei, P.; Izsvák, Z.; Narayanavari, S.A.; Singh, H.; Ivics, Z. Gene Therapy with the Sleeping Beauty Transposon System. Trends Genet. 2017, 33, 852–870. [Google Scholar] [CrossRef] [PubMed]
- Bulcha, J.T.; Wang, Y.; Ma, H.; Tai, P.W.L.; Gao, G. Viral Vector Platforms within the Gene Therapy Landscape. Sig. Transduct. Target Ther. 2021, 6, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Hagedorn, C.; Wong, S.-P.; Harbottle, R.; Lipps, H.J. Scaffold/Matrix Attached Region-Based Nonviral Episomal Vectors. Hum. Gene Ther. 2011, 22, 915–923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weklak, D.; Pembaur, D.; Koukou, G.; Jönsson, F.; Hagedorn, C.; Kreppel, F. Genetic and Chemical Capsid Modifications of Adenovirus Vectors to Modulate Vector–Host Interactions. Viruses 2021, 13, 1300. [Google Scholar] [CrossRef] [PubMed]
- Russell, S.; Bennett, J.; Wellman, J.A.; Chung, D.C.; Yu, Z.-F.; Tillman, A.; Wittes, J.; Pappas, J.; Elci, O.; McCague, S.; et al. Efficacy and Safety of Voretigene Neparvovec (AAV2-HRPE65v2) in Patients with RPE65-Mediated Inherited Retinal Dystrophy: A Randomised, Controlled, Open-Label, Phase 3 Trial. Lancet 2017, 390, 849–860. [Google Scholar] [CrossRef]
- Day, J.W.; Finkel, R.S.; Chiriboga, C.A.; Connolly, A.M.; Crawford, T.O.; Darras, B.T.; Iannaccone, S.T.; Kuntz, N.L.; Peña, L.D.M.; Shieh, P.B.; et al. Onasemnogene Abeparvovec Gene Therapy for Symptomatic Infantile-Onset Spinal Muscular Atrophy in Patients with Two Copies of SMN2 (STR1VE): An Open-Label, Single-Arm, Multicentre, Phase 3 Trial. Lancet Neurol. 2021, 20, 284–293. [Google Scholar] [CrossRef]
- High-Dose AAV Gene Therapy Deaths. Nat. Biotechnol. 2020, 38, 910. [CrossRef] [PubMed]
- Somanathan, S.; Calcedo, R.; Wilson, J.M. Adenovirus-Antibody Complexes Contributed to Lethal Systemic Inflammation in a Gene Therapy Trial. Mol. Ther. 2020, 28, 784–793. [Google Scholar] [CrossRef] [PubMed]
- Folegatti, P.M.; Ewer, K.J.; Aley, P.K.; Angus, B.; Becker, S.; Belij-Rammerstorfer, S.; Bellamy, D.; Bibi, S.; Bittaye, M.; Clutterbuck, E.A.; et al. Safety and Immunogenicity of the ChAdOx1 NCoV-19 Vaccine against SARS-CoV-2: A Preliminary Report of a Phase 1/2, Single-Blind, Randomised Controlled Trial. Lancet 2020, 396, 467–478. [Google Scholar] [CrossRef]
- Khuri, F.R.; Nemunaitis, J.; Ganly, I.; Arseneau, J.; Tannock, I.F.; Romel, L.; Gore, M.; Ironside, J.; MacDougall, R.H.; Heise, C.; et al. A Controlled Trial of Intratumoral ONYX-015, a Selectively-Replicating Adenovirus, in Combination with Cisplatin and 5-Fluorouracil in Patients with Recurrent Head and Neck Cancer. Nat. Med. 2000, 6, 879–885. [Google Scholar] [CrossRef] [PubMed]
- Dalwadi, D.A.; Calabria, A.; Tiyaboonchai, A.; Posey, J.; Naugler, W.E.; Montini, E.; Grompe, M. AAV Integration in Human Hepatocytes. Mol. Ther. 2021, 29, 2898–2909. [Google Scholar] [CrossRef] [PubMed]
- Hacein-Bey-Abina, S.; Garrigue, A.; Wang, G.P.; Soulier, J.; Lim, A.; Morillon, E.; Clappier, E.; Caccavelli, L.; Delabesse, E.; Beldjord, K.; et al. Insertional Oncogenesis in 4 Patients after Retrovirus-Mediated Gene Therapy of SCID-X1. J. Clin. Investig. 2008, 118, 3132–3142. [Google Scholar] [CrossRef] [PubMed]
- Hacein-Bey-Abina, S.; Von Kalle, C.; Schmidt, M.; McCormack, M.P.; Wulffraat, N.; Leboulch, P.; Lim, A.; Osborne, C.S.; Pawliuk, R.; Morillon, E.; et al. LMO2-Associated Clonal T Cell Proliferation in Two Patients after Gene Therapy for SCID-X1. Science 2003, 302, 415–419. [Google Scholar] [CrossRef] [PubMed]
- Howe, S.J.; Mansour, M.R.; Schwarzwaelder, K.; Bartholomae, C.; Hubank, M.; Kempski, H.; Brugman, M.H.; Pike-Overzet, K.; Chatters, S.J.; De Ridder, D.; et al. Insertional Mutagenesis Combined with Acquired Somatic Mutations Causes Leukemogenesis Following Gene Therapy of SCID-X1 Patients. J. Clin. Investig. 2008, 118, 3143–3150. [Google Scholar] [CrossRef] [PubMed]
- Dougherty, J.P.; Temin, H.M. A Promoterless Retroviral Vector Indicates That There Are Sequences in U3 Required for 3’ RNA Processing. Proc. Natl. Acad. Sci. USA 1987, 84, 1197–1201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hawley, R.G.; Covarrubias, L.; Hawley, T.; Mintz, B. Handicapped Retroviral Vectors Efficiently Transduce Foreign Genes into Hematopoietic Stem Cells. Proc. Natl. Acad. Sci. USA 1987, 84, 2406–2410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, S.F.; von Rüden, T.; Kantoff, P.W.; Garber, C.; Seiberg, M.; Rüther, U.; Anderson, W.F.; Wagner, E.F.; Gilboa, E. Self-Inactivating Retroviral Vectors Designed for Transfer of Whole Genes into Mammalian Cells. Proc. Natl. Acad. Sci. USA 1986, 83, 3194–3198. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.L.; Li, H.; Wang, Y.; Zhu, F.X.; Kudchodkar, S.; Yuan, Y. Kaposi’s Sarcoma-Associated Herpesvirus Lytic Origin (Ori-Lyt)-Dependent DNA Replication: Identification of the Ori-Lyt and Association of K8 BZip Protein with the Origin. J. Virol. 2003, 77, 5578–5588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsurumi, T.; Fujita, M.; Kudoh, A. Latent and Lytic Epstein-Barr Virus Replication Strategies. Rev. Med. Virol. 2005, 15, 3–15. [Google Scholar] [CrossRef] [PubMed]
- Rose, C.; Green, M.; Webber, S.; Kingsley, L.; Day, R.; Watkins, S.; Reyes, J.; Rowe, D. Detection of Epstein-Barr Virus Genomes in Peripheral Blood B Cells from Solid-Organ Transplant Recipients by Fluorescence in Situ Hybridization. J. Clin. Microbiol. 2002, 40, 2533–2544. [Google Scholar] [CrossRef] [Green Version]
- Reisman, D.; Yates, J.; Sugden, B. A Putative Origin of Replication of Plasmids Derived from Epstein-Barr Virus Is Composed of Two Cis-Acting Components. Mol. Cell. Biol. 1985, 5, 1822–1832. [Google Scholar]
- Shirakata, M.; Hirai, K. Identification of Minimal OriP of Epstein-Barr Virus Required for DNA Replication. J. Biochem. 1998, 123, 175–181. [Google Scholar] [CrossRef] [PubMed]
- Yates, J.L.; Camiolo, S.M.; Bashaw, J.M. The Minimal Replicator of Epstein-Barr Virus OriP. J. Virol. 2000, 74, 4512–4522. [Google Scholar] [CrossRef] [PubMed]
- Kempkes, B.; Pich, D.; Zeidler, R.; Hammerschmidt, W. Immortalization of Human Primary B Lymphocytes in Vitro with DNA. Proc. Natl. Acad. Sci. USA 1995, 92, 5875–5879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaye, K.M.; Izumi, K.M.; Li, H.; Johannsen, E.; Davidson, D.; Longnecker, R.; Kieff, E. An Epstein-Barr Virus That Expresses Only the First 231 LMP1 Amino Acids Efficiently Initiates Primary B-Lymphocyte Growth Transformation. J. Virol. 1999, 73, 10525–10530. [Google Scholar] [CrossRef] [Green Version]
- Humme, S.; Reisbach, G.; Feederle, R.; Delecluse, H.J.; Bousset, K.; Hammerschmidt, W.; Schepers, A. The EBV Nuclear Antigen 1 (EBNA1) Enhances B Cell Immortalization Several Thousandfold. Proc. Natl. Acad. Sci. USA 2003, 100, 10989–10994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Black, J.; Vos, J.-M. Establishment of an OriP/EBNA1-Based Episomal Vector Transcribing Human Genomic Beta-Globin in Cultured Murine Fibroblasts. Gene Ther. 2002, 9, 1447–1454. [Google Scholar] [CrossRef] [PubMed]
- Hung, S.C.; Kang, M.S.; Kieff, E. Maintenance of Epstein-Barr Virus (EBV) OriP-Based Episomes Requires EBV-Encoded Nuclear Antigen-1 Chromosome-Binding Domains, Which Can Be Replaced by High-Mobility Group-I or Histone H1. Proc. Natl. Acad. Sci. USA 2001, 98, 1865–1870. [Google Scholar] [CrossRef] [PubMed]
- Sears, J.; Ujihara, M.; Wong, S.; Ott, C.; Middeldorp, J.; Aiyar, A. The Amino Terminus of Epstein-Barr Virus (EBV) Nuclear Antigen 1 Contains AT Hooks That Facilitate the Replication and Partitioning of Latent EBV Genomes by Tethering Them to Cellular Chromosomes. J. Virol. 2004, 78, 11487–11505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomae, A.W.; Pich, D.; Brocher, J.; Spindler, M.-P.; Berens, C.; Hock, R.; Hammerschmidt, W.; Schepers, A. Interaction between HMGA1a and the Origin Recognition Complex Creates Site-Specific Replication Origins. Proc. Natl. Acad. Sci. USA 2008, 105, 1692–1697. [Google Scholar] [CrossRef] [PubMed]
- Ivics, Z.; Hackett, P.B.; Plasterk, R.H.; Izsvák, Z. Molecular Reconstruction of Sleeping Beauty, a Tc1-like Transposon from Fish, and Its Transposition in Human Cells. Cell 1997, 91, 501–510. [Google Scholar] [CrossRef] [Green Version]
- Finnegan, D.J. Eukaryotic Transposable Elements and Genome Evolution. Trends Genet. 1989, 5, 103–107. [Google Scholar] [CrossRef]
- Van Luenen, H.G.; Colloms, S.D.; Plasterk, R.H. The Mechanism of Transposition of Tc3 in C. Elegans. Cell 1994, 79, 293–301. [Google Scholar] [CrossRef]
- Plasterk, R.H.; Izsvák, Z.; Ivics, Z. Resident Aliens: The Tc1/Mariner Superfamily of Transposable Elements. Trends Genet. 1999, 15, 326–332. [Google Scholar] [CrossRef]
- Holstein, M.; Mesa-Nuñez, C.; Miskey, C.; Almarza, E.; Poletti, V.; Schmeer, M.; Grueso, E.; Ordóñez Flores, J.C.; Kobelt, D.; Walther, W.; et al. Efficient Non-Viral Gene Delivery into Human Hematopoietic Stem Cells by Minicircle Sleeping Beauty Transposon Vectors. Mol. Ther. 2018, 26, 1137–1153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilber, A.; Frandsen, J.L.; Geurts, J.L.; Largaespada, D.A.; Hackett, P.B.; McIvor, R.S. RNA as a Source of Transposase for Sleeping Beauty-Mediated Gene Insertion and Expression in Somatic Cells and Tissues. Mol. Ther. 2006, 13, 625–630. [Google Scholar] [CrossRef] [PubMed]
- Querques, I.; Mades, A.; Zuliani, C.; Miskey, C.; Alb, M.; Grueso, E.; Machwirth, M.; Rausch, T.; Einsele, H.; Ivics, Z.; et al. A Highly Soluble Sleeping Beauty Transposase Improves Control of Gene Insertion. Nat. Biotechnol. 2019, 37, 1502–1512. [Google Scholar] [CrossRef] [PubMed]
- Alhaji, S.Y.; Ngai, S.C.; Abdullah, S. Silencing of Transgene Expression in Mammalian Cells by DNA Methylation and Histone Modifications in Gene Therapy Perspective. Biotechnol. Genet. Eng. Rev. 2019, 35, 1–25. [Google Scholar] [CrossRef]
- Boyes, J.; Bird, A. Repression of Genes by DNA Methylation Depends on CpG Density and Promoter Strength: Evidence for Involvement of a Methyl-CpG Binding Protein. EMBO J. 1992, 11, 327–333. [Google Scholar] [CrossRef] [PubMed]
- Illingworth, R.S.; Bird, A.P. CpG Islands—’A Rough Guide’. FEBS Lett. 2009, 583, 1713–1720. [Google Scholar] [CrossRef] [PubMed]
- Piechaczek, C.; Fetzer, C.; Baiker, A.; Bode, J.; Lipps, H.J. A Vector Based on the SV40 Origin of Replication and Chromosomal S/MARs Replicates Episomally in CHO Cells. Nucleic Acids Res. 1999, 27, 426–428. [Google Scholar] [CrossRef] [PubMed]
- Papapetrou, E.P.; Ziros, P.G.; Micheva, I.D.; Zoumbos, N.C.; Athanassiadou, A. Gene Transfer into Human Hematopoietic Progenitor Cells with an Episomal Vector Carrying an S/MAR Element. Gene Ther. 2006, 13, 40–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stehle, I.M.; Scinteie, M.F.; Baiker, A.; Jenke, A.C.; Lipps, H.J. Exploiting a Minimal System to Study the Epigenetic Control of DNA Replication: The Interplay between Transcription and Replication. Chromosome Res. 2003, 11, 413–421. [Google Scholar] [CrossRef] [PubMed]
- Schaarschmidt, D.; Baltin, J.; Stehle, I.M.; Lipps, H.J.; Knippers, R. An Episomal Mammalian Replicon: Sequence-Independent Binding of the Origin Recognition Complex. EMBO J. 2004, 23, 191–201. [Google Scholar] [CrossRef] [Green Version]
- Stehle, I.M.; Postberg, J.; Rupprecht, S.; Cremer, T.; Jackson, D.A.; Lipps, H.J. Establishment and Mitotic Stability of an Extra-Chromosomal Mammalian Replicon. BMC Cell Biol. 2007, 8, 33. [Google Scholar] [CrossRef] [Green Version]
- Harrington, J.J.; Van Bokkelen, G.; Mays, R.W.; Gustashaw, K.; Willard, H.F. Formation of de Novo Centromeres and Construction of First-Generation Human Artificial Microchromosomes. Nat. Genet. 1997, 15, 345–355. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, H.; Singer, M.; Rosenberg, M. Highly Reiterated Sequences of SIMIANSIMIANSIMIANSIMIANSIMIAN. Science 1978, 200, 394–402. [Google Scholar] [CrossRef] [PubMed]
- Thayer, R.E.; Singer, M.F.; McCutchan, T.F. Sequence Relationships between Single Repeat Units of Highly Reiterated African Green Monkey DNA. Nucleic Acids Res. 1981, 9, 169–181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mejia, J.E.; Willmott, A.; Levy, E.; Earnshaw, W.C.; Larin, Z. Functional Complementation of a Genetic Deficiency with Human Artificial Chromosomes. Am. J. Hum. Genet. 2001, 69, 315–326. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.W.; Pendon, C.; Yang, J.; Haywood, N.; Chand, A.; Brown, W.R. Human Mini-Chromosomes with Minimal Centromeres. Hum. Mol. Genet. 2000, 9, 1891–1902. [Google Scholar] [CrossRef] [PubMed]
- Lo, A.W.; Liao, G.C.; Rocchi, M.; Choo, K.H. Extreme Reduction of Chromosome-Specific Alpha-Satellite Array Is Unusually Common in Human Chromosome 21. Genome Res. 1999, 9, 895–908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kotzamanis, G.; Cheung, W.; Abdulrazzak, H.; Perez-Luz, S.; Howe, S.; Cooke, H.; Huxley, C. Construction of Human Artificial Chromosome Vectors by Recombineering. Gene 2005, 351, 29–38. [Google Scholar] [CrossRef] [PubMed]
- Navaratnarajah, C.K.; Leonard, V.H.J.; Cattaneo, R. Measles Virus Glycoprotein Complex Assembly, Receptor Attachment, and Cell Entry. Curr. Top. Microbiol. Immunol. 2009, 329, 59–76. [Google Scholar] [CrossRef] [PubMed]
- Frenkel, N. The History of the HSV Amplicon: From Naturally Occurring Defective Genomes to Engineered Amplicon Vectors. Curr. Gene Ther. 2006, 6, 277–301. [Google Scholar] [CrossRef] [PubMed]
- Ponomartsev, S.V.; Sinenko, S.A.; Skvortsova, E.V.; Liskovykh, M.A.; Voropaev, I.N.; Savina, M.M.; Kuzmin, A.A.; Kuzmina, E.Y.; Kondrashkina, A.M.; Larionov, V.; et al. Human AlphoidtetO Artificial Chromosome as a Gene Therapy Vector for the Developing Hemophilia A Model in Mice. Cells 2020, 9, 879. [Google Scholar] [CrossRef] [Green Version]
- Chan, D.Y.; Moralli, D.; Wheatley, L.; Jankowska, J.D.; Monaco, Z.L. Multigene Human Artificial Chromosome Vector Delivery with Herpes Simplex Virus 1 Amplicons. Exp. Cell Res. 2020, 388, 111840. [Google Scholar] [CrossRef]
- Kouprina, N.; Earnshaw, W.C.; Masumoto, H.; Larionov, V. A New Generation of Human Artificial Chromosomes for Functional Genomics and Gene Therapy. Cell Mol. Life Sci. 2013, 70, 1135–1148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kouprina, N.; Petrov, N.; Molina, O.; Liskovykh, M.; Pesenti, E.; Ohzeki, J.-I.; Masumoto, H.; Earnshaw, W.C.; Larionov, V. Human Artificial Chromosome with Regulated Centromere: A Tool for Genome and Cancer Studies. ACS Synth. Biol. 2018, 7, 1974–1989. [Google Scholar] [CrossRef]
- Ohzeki, J.-I.; Otake, K.; Masumoto, H. Human Artificial Chromosome: Chromatin Assembly Mechanisms and CENP-B. Exp. Cell Res. 2020, 389, 111900. [Google Scholar] [CrossRef]
- Moralli, D.; Monaco, Z.L. Gene Expressing Human Artificial Chromosome Vectors: Advantages and Challenges for Gene Therapy. Exp. Cell Res. 2020, 390, 111931. [Google Scholar] [CrossRef] [PubMed]
- Ikeno, M.; Hasegawa, Y. Applications of Bottom-up Human Artificial Chromosomes in Cell Research and Cell Engineering. Exp. Cell Res. 2020, 390, 111793. [Google Scholar] [CrossRef] [PubMed]
- Foecking, M.K.; Hofstetter, H. Powerful and Versatile Enhancer-Promoter Unit for Mammalian Expression Vectors. Gene 1986, 45, 101–105. [Google Scholar] [CrossRef]
- Xia, W.; Bringmann, P.; McClary, J.; Jones, P.P.; Manzana, W.; Zhu, Y.; Wang, S.; Liu, Y.; Harvey, S.; Madlansacay, M.R.; et al. High Levels of Protein Expression Using Different Mammalian CMV Promoters in Several Cell Lines. Protein Expr. Purif. 2006, 45, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Sakurai, F.; Kawabata, K.; Yamaguchi, T.; Hayakawa, T.; Mizuguchi, H. Optimization of Adenovirus Serotype 35 Vectors for Efficient Transduction in Human Hematopoietic Progenitors: Comparison of Promoter Activities. Gene Ther. 2005, 12, 1424–1433. [Google Scholar] [CrossRef] [Green Version]
- Manzini, S.; Vargiolu, A.; Stehle, I.M.; Bacci, M.L.; Cerrito, M.G.; Giovannoni, R.; Zannoni, A.; Bianco, M.R.; Forni, M.; Donini, P.; et al. Genetically Modified Pigs Produced with a Nonviral Episomal Vector. Proc. Natl. Acad. Sci. USA 2006, 103, 17672–17677. [Google Scholar] [CrossRef] [Green Version]
- Hagedorn, C.; Gogol-Döring, A.; Schreiber, S.; Epplen, J.T.; Lipps, H.J. Genome-Wide Profiling of S/MAR-Based Replicon Contact Sites. Nucleic Acids Res. 2017, 45, 7841–7854. [Google Scholar] [CrossRef] [Green Version]
- Qin, J.Y.; Zhang, L.; Clift, K.L.; Hulur, I.; Xiang, A.P.; Ren, B.-Z.; Lahn, B.T. Systematic Comparison of Constitutive Promoters and the Doxycycline-Inducible Promoter. PLoS ONE 2010, 5, e10611. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Krohn, J.; Bhattacharya, S.; Davies, B. A Comparison of Exogenous Promoter Activity at the ROSA26 Locus Using a ΦiC31 Integrase Mediated Cassette Exchange Approach in Mouse ES Cells. PLoS ONE 2011, 6, e23376. [Google Scholar] [CrossRef] [Green Version]
- Haase, R.; Argyros, O.; Wong, S.-P.; Harbottle, R.P.; Lipps, H.J.; Ogris, M.; Magnusson, T.; Vizoso Pinto, M.G.; Haas, J.; Baiker, A. PEPito: A Significantly Improved Non-Viral Episomal Expression Vector for Mammalian Cells. BMC Biotechnol. 2010, 10, 20. [Google Scholar] [CrossRef] [Green Version]
- Sawicki, J.A.; Morris, R.J.; Monks, B.; Sakai, K.; Miyazaki, J. A Composite CMV-IE Enhancer/Beta-Actin Promoter Is Ubiquitously Expressed in Mouse Cutaneous Epithelium. Exp. Cell Res. 1998, 244, 367–369. [Google Scholar] [CrossRef] [PubMed]
- Drozd, A.M.; Walczak, M.P.; Piaskowski, S.; Stoczynska-Fidelus, E.; Rieske, P.; Grzela, D.P. Generation of Human IPSCs from Cells of Fibroblastic and Epithelial Origin by Means of the OriP/EBNA-1 Episomal Reprogramming System. Stem Cell Res. Ther. 2015, 6, 122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roig-Merino, A.; Urban, M.; Bozza, M.; Peterson, J.D.; Bullen, L.; Büchler-Schäff, M.; Stäble, S.; van der Hoeven, F.; Müller-Decker, K.; McKay, T.R.; et al. An Episomal DNA Vector Platform for the Persistent Genetic Modification of Pluripotent Stem Cells and Their Differentiated Progeny. Stem Cell Rep. 2022, 17, 143–158. [Google Scholar] [CrossRef] [PubMed]
- Khabou, H.; Cordeau, C.; Pacot, L.; Fisson, S.; Dalkara, D. Dosage Thresholds and Influence of Transgene Cassette in Adeno-Associated Virus-Related Toxicity. Hum. Gene Ther. 2018, 29, 1235–1241. [Google Scholar] [CrossRef]
- Bozza, M.; Roia, A.D.; Correia, M.P.; Berger, A.; Tuch, A.; Schmidt, A.; Zörnig, I.; Jäger, D.; Schmidt, P.; Harbottle, R.P. A Nonviral, Nonintegrating DNA Nanovector Platform for the Safe, Rapid, and Persistent Manufacture of Recombinant T Cells. Sci. Adv. 2021, 7, eabf1333. [Google Scholar] [CrossRef]
- Thyagarajan, B.; Scheyhing, K.; Xue, H.; Fontes, A.; Chesnut, J.; Rao, M.; Lakshmipathy, U. A Single EBV-Based Vector for Stable Episomal Maintenance and Expression of GFP in Human Embryonic Stem Cells. Regen. Med. 2009, 4, 239–250. [Google Scholar] [CrossRef]
- Argyros, O.; Wong, S.P.; Fedonidis, C.; Tolmachov, O.; Waddington, S.N.; Howe, S.J.; Niceta, M.; Coutelle, C.; Harbottle, R.P. Development of S/MAR Minicircles for Enhanced and Persistent Transgene Expression in the Mouse Liver. J. Mol. Med. 2011, 89, 515–529. [Google Scholar] [CrossRef]
- Haase, R.; Magnusson, T.; Su, B.; Kopp, F.; Wagner, E.; Lipps, H.; Baiker, A.; Ogris, M. Generation of a Tumor- and Tissue-Specific Episomal Non-Viral Vector System. BMC Biotechnol. 2013, 13, 49. [Google Scholar] [CrossRef] [Green Version]
- Dalsgaard, T.; Moldt, B.; Sharma, N.; Wolf, G.; Schmitz, A.; Pedersen, F.S.; Mikkelsen, J.G. Shielding of Sleeping Beauty DNA Transposon-Delivered Transgene Cassettes by Heterologous Insulators in Early Embryonal Cells. Mol. Ther. 2009, 17, 121–130. [Google Scholar] [CrossRef]
- Arumugam, P.I.; Scholes, J.; Perelman, N.; Xia, P.; Yee, J.-K.; Malik, P. Improved Human Beta-Globin Expression from Self-Inactivating Lentiviral Vectors Carrying the Chicken Hypersensitive Site-4 (CHS4) Insulator Element. Mol. Ther. 2007, 15, 1863–1871. [Google Scholar] [CrossRef]
- Hagedorn, C.; Antoniou, M.N.; Lipps, H.J. Genomic Cis-Acting Sequences Improve Expression and Establishment of a Nonviral Vector. Mol. Ther. Nucleic Acids 2013, 2, e118. [Google Scholar] [CrossRef] [PubMed]
- Skipper, K.A.; Hollensen, A.K.; Antoniou, M.N.; Mikkelsen, J.G. Sustained Transgene Expression from Sleeping Beauty DNA Transposons Containing a Core Fragment of the HNRPA2B1-CBX3 Ubiquitous Chromatin Opening Element (UCOE). BMC Biotechnol. 2019, 19, 75. [Google Scholar] [CrossRef] [PubMed]
- Sjeklocha, L.; Chen, Y.; Daly, M.C.; Steer, C.J.; Kren, B.T. β-Globin Matrix Attachment Region Improves Stable Genomic Expression of the Sleeping Beauty Transposon. J. Cell Biochem. 2011, 112, 2361–2375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saunders, F.; Sweeney, B.; Antoniou, M.N.; Stephens, P.; Cain, K. Chromatin Function Modifying Elements in an Industrial Antibody Production Platform - Comparison of UCOE, MAR, STAR and CHS4 Elements. PLoS ONE 2015, 10, e0120096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antoniou, M.; Harland, L.; Mustoe, T.; Williams, S.; Holdstock, J.; Yague, E.; Mulcahy, T.; Griffiths, M.; Edwards, S.; Ioannou, P.A.; et al. Transgenes Encompassing Dual-Promoter CpG Islands from the Human TBP and HNRPA2B1 Loci Are Resistant to Heterochromatin-Mediated Silencing. Genomics 2003, 82, 269–279. [Google Scholar] [CrossRef]
- Williams, S.; Mustoe, T.; Mulcahy, T.; Griffiths, M.; Simpson, D.; Antoniou, M.; Irvine, A.; Mountain, A.; Crombie, R. CpG-Island Fragments from the HNRPA2B1/CBX3 Genomic Locus Reduce Silencing and Enhance Transgene Expression from the HCMV Promoter/Enhancer in Mammalian Cells. BMC Biotechnol. 2005, 5, 17. [Google Scholar] [CrossRef] [Green Version]
- Müller-Kuller, U.; Ackermann, M.; Kolodziej, S.; Brendel, C.; Fritsch, J.; Lachmann, N.; Kunkel, H.; Lausen, J.; Schambach, A.; Moritz, T.; et al. A Minimal Ubiquitous Chromatin Opening Element (UCOE) Effectively Prevents Silencing of Juxtaposed Heterologous Promoters by Epigenetic Remodeling in Multipotent and Pluripotent Stem Cells. Nucleic Acids Res. 2015, 43, 1577–1592. [Google Scholar] [CrossRef] [Green Version]
- Zhang, F.; Santilli, G.; Thrasher, A.J. Characterization of a Core Region in the A2UCOE That Confers Effective Anti-Silencing Activity. Sci. Rep. 2017, 7, 10213. [Google Scholar] [CrossRef] [Green Version]
- Bell, A.C.; West, A.G.; Felsenfeld, G. The Protein CTCF Is Required for the Enhancer Blocking Activity of Vertebrate Insulators. Cell 1999, 98, 387–396. [Google Scholar] [CrossRef] [Green Version]
- Aker, M.; Bomsztyk, K.; Emery, D.W. Poly(ADP-Ribose) Polymerase-1 (PARP-1) Contributes to the Barrier Function of a Vertebrate Chromatin Insulator. J. Biol. Chem. 2010, 285, 37589–37597. [Google Scholar] [CrossRef] [Green Version]
- West, A.G.; Huang, S.; Gaszner, M.; Litt, M.D.; Felsenfeld, G. Recruitment of Histone Modifications by USF Proteins at a Vertebrate Barrier Element. Mol. Cell 2004, 16, 453–463. [Google Scholar] [CrossRef] [PubMed]
- Gerasimova, T.I.; Byrd, K.; Corces, V.G. A Chromatin Insulator Determines the Nuclear Localization of DNA. Mol. Cell 2000, 6, 1025–1035. [Google Scholar] [CrossRef]
- Yusufzai, T.M.; Tagami, H.; Nakatani, Y.; Felsenfeld, G. CTCF Tethers an Insulator to Subnuclear Sites, Suggesting Shared Insulator Mechanisms across Species. Mol. Cell 2004, 13, 291–298. [Google Scholar] [CrossRef]
- Little, R.D.; Schildkraut, C.L. Initiation of Latent DNA Replication in the Epstein-Barr Virus Genome Can Occur at Sites Other than the Genetically Defined Origin. Mol. Cell Biol. 1995, 15, 2893–2903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Norio, P.; Schildkraut, C.L.; Yates, J.L. Initiation of DNA Replication within OriP Is Dispensable for Stable Replication of the Latent Epstein-Barr Virus Chromosome after Infection of Established Cell Lines. J. Virol. 2000, 74, 8563–8574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ott, E.; Norio, P.; Ritzi, M.; Schildkraut, C.; Schepers, A. The Dyad Symmetry Element of Epstein-Barr Virus Is a Dominant but Dispensable Replication Origin. PLoS ONE 2011, 6, e18609. [Google Scholar] [CrossRef] [PubMed]
- Boulikas, T. Common Structural Features of Replication Origins in All Life Forms. J. Cell Biochem. 1996, 60, 297–316. [Google Scholar] [CrossRef]
- Gerhardt, J.; Jafar, S.; Spindler, M.-P.; Ott, E.; Schepers, A. Identification of New Human Origins of DNA Replication by an Origin-Trapping Assay. Mol. Cell Biol. 2006, 26, 7731–7746. [Google Scholar] [CrossRef] [Green Version]
- Reeves, R.; Nissen, M.S. The A.T-DNA-Binding Domain of Mammalian High Mobility Group I Chromosomal Proteins. A Novel Peptide Motif for Recognizing DNA Structure. J. Biol. Chem. 1990, 265, 8573–8582. [Google Scholar] [CrossRef]
- Pich, D.; Humme, S.; Spindler, M.-P.; Schepers, A.; Hammerschmidt, W. Conditional Gene Vectors Regulated in Cis. Nucleic Acids Res. 2008, 36, e83. [Google Scholar] [CrossRef] [Green Version]
- Baiker, A.; Maercker, C.; Piechaczek, C.; Schmidt, S.B.; Bode, J.; Benham, C.; Lipps, H.J. Mitotic Stability of an Episomal Vector Containing a Human Scaffold/Matrix-Attached Region Is Provided by Association with Nuclear Matrix. Nat. Cell Biol. 2000, 2, 182–184. [Google Scholar] [CrossRef] [PubMed]
- Jenke, A.C.; Stehle, I.M.; Herrmann, F.; Eisenberger, T.; Baiker, A.; Bode, J.; Fackelmayer, F.O.; Lipps, H.J. Nuclear Scaffold/Matrix Attached Region Modules Linked to a Transcription Unit Are Sufficient for Replication and Maintenance of a Mammalian Episome. Proc. Natl. Acad. Sci. USA 2004, 101, 11322–11327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rupprecht, S.; Hagedorn, C.; Seruggia, D.; Magnusson, T.; Wagner, E.; Ogris, M.; Lipps, H.J. Controlled Removal of a Nonviral Episomal Vector from Transfected Cells. Gene 2010, 466, 36–42. [Google Scholar] [CrossRef] [PubMed]
- Stavrou, E.F.; Lazaris, V.M.; Giannakopoulos, A.; Papapetrou, E.; Spyridonidis, A.; Zoumbos, N.C.; Gkountis, A.; Athanassiadou, A. The β-Globin Replicator Greatly Enhances the Potential of S/MAR Based Episomal Vectors for Gene Transfer into Human Haematopoietic Progenitor Cells. Sci. Rep. 2017, 7, 40673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stavrou, E.F.; Simantirakis, E.; Verras, M.; Barbas, C.; Vassilopoulos, G.; Peterson, K.R.; Athanassiadou, A. Episomal Vectors Based on S/MAR and the β-Globin Replicator, Encoding a Synthetic Transcriptional Activator, Mediate Efficient γ-Globin Activation in Haematopoietic Cells. Sci. Rep. 2019, 9, 19765. [Google Scholar] [CrossRef] [Green Version]
- Valton, A.-L.; Prioleau, M.-N. G-Quadruplexes in DNA Replication: A Problem or a Necessity? Trends Genet. 2016, 32, 697–706. [Google Scholar] [CrossRef]
- Rhodes, D.; Lipps, H.J. G-Quadruplexes and Their Regulatory Roles in Biology. Nucleic Acids Res. 2015, 43, 8627–8637. [Google Scholar] [CrossRef] [Green Version]
- Cayrou, C.; Coulombe, P.; Vigneron, A.; Stanojcic, S.; Ganier, O.; Peiffer, I.; Rivals, E.; Puy, A.; Laurent-Chabalier, S.; Desprat, R.; et al. Genome-Scale Analysis of Metazoan Replication Origins Reveals Their Organization in Specific but Flexible Sites Defined by Conserved Features. Genome Res. 2011, 21, 1438–1449. [Google Scholar] [CrossRef] [Green Version]
- Comoglio, F.; Schlumpf, T.; Schmid, V.; Rohs, R.; Beisel, C.; Paro, R. High-Resolution Profiling of Drosophila Replication Start Sites Reveals a DNA Shape and Chromatin Signature of Metazoan Origins. Cell Rep. 2015, 11, 821–834. [Google Scholar] [CrossRef] [Green Version]
- Cayrou, C.; Ballester, B.; Peiffer, I.; Fenouil, R.; Coulombe, P.; Andrau, J.-C.; van Helden, J.; Méchali, M. The Chromatin Environment Shapes DNA Replication Origin Organization and Defines Origin Classes. Genome Res. 2015, 25, 1873–1885. [Google Scholar] [CrossRef] [Green Version]
- Prorok, P.; Artufel, M.; Aze, A.; Coulombe, P.; Peiffer, I.; Lacroix, L.; Guédin, A.; Mergny, J.-L.; Damaschke, J.; Schepers, A.; et al. Involvement of G-Quadruplex Regions in Mammalian Replication Origin Activity. Nat. Commun. 2019, 10, 3274. [Google Scholar] [CrossRef] [PubMed]
- Norseen, J.; Thomae, A.; Sridharan, V.; Aiyar, A.; Schepers, A.; Lieberman, P.M. RNA-Dependent Recruitment of the Origin Recognition Complex. EMBO J. 2008, 27, 3024–3035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Norseen, J.; Johnson, F.B.; Lieberman, P.M. Role for G-Quadruplex RNA Binding by Epstein-Barr Virus Nuclear Antigen 1 in DNA Replication and Metaphase Chromosome Attachment. J. Virol. 2009, 83, 10336–10346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimizu, T.S.; Takahashi, K.; Tomita, M. CpG Distribution Patterns in Methylated and Non-Methylated Species. Gene 1997, 205, 103–107. [Google Scholar] [CrossRef]
- Ishii, K.J.; Akira, S. Innate Immune Recognition of, and Regulation by, DNA. Trends Immunol. 2006, 27, 525–532. [Google Scholar] [CrossRef] [PubMed]
- Bauer, S.; Kirschning, C.J.; Häcker, H.; Redecke, V.; Hausmann, S.; Akira, S.; Wagner, H.; Lipford, G.B. Human TLR9 Confers Responsiveness to Bacterial DNA via Species-Specific CpG Motif Recognition. Proc. Natl. Acad. Sci. USA 2001, 98, 9237–9242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hyde, S.C.; Pringle, I.A.; Abdullah, S.; Lawton, A.E.; Davies, L.A.; Varathalingam, A.; Nunez-Alonso, G.; Green, A.-M.; Bazzani, R.P.; Sumner-Jones, S.G.; et al. CpG-Free Plasmids Confer Reduced Inflammation and Sustained Pulmonary Gene Expression. Nat. Biotechnol. 2008, 26, 549–551. [Google Scholar] [CrossRef] [Green Version]
- Yew, N.S.; Zhao, H.; Przybylska, M.; Wu, I.-H.; Tousignant, J.D.; Scheule, R.K.; Cheng, S.H. CpG-Depleted Plasmid DNA Vectors with Enhanced Safety and Long-Term Gene Expression in Vivo. Mol. Ther. 2002, 5, 731–738. [Google Scholar] [CrossRef]
- Darquet, A.M.; Rangara, R.; Kreiss, P.; Schwartz, B.; Naimi, S.; Delaère, P.; Crouzet, J.; Scherman, D. Minicircle: An Improved DNA Molecule for in Vitro and in Vivo Gene Transfer. Gene Ther. 1999, 6, 209–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Broll, S.; Oumard, A.; Hahn, K.; Schambach, A.; Bode, J. Minicircle Performance Depending on S/MAR-Nuclear Matrix Interactions. J. Mol. Biol. 2010, 395, 950–965. [Google Scholar] [CrossRef]
- Luke, J.M.; Vincent, J.M.; Du, S.X.; Gerdemann, U.; Leen, A.M.; Whalen, R.G.; Hodgson, C.P.; Williams, J.A. Improved Antibiotic-Free Plasmid Vector Design by Incorporation of Transient Expression Enhancers. Gene Ther. 2011, 18, 334–343. [Google Scholar] [CrossRef] [PubMed]
- Marie, C.; Vandermeulen, G.; Quiviger, M.; Richard, M.; Préat, V.; Scherman, D. PFARs, Plasmids Free of Antibiotic Resistance Markers, Display High-Level Transgene Expression in Muscle, Skin and Tumour Cells. J. Gene Med. 2010, 12, 323–332. [Google Scholar] [CrossRef] [PubMed]
- Bozza, M.; Green, E.W.; Espinet, E.; De Roia, A.; Klein, C.; Vogel, V.; Offringa, R.; Williams, J.A.; Sprick, M.; Harbottle, R.P. Novel Non-Integrating DNA Nano-S/MAR Vectors Restore Gene Function in Isogenic Patient-Derived Pancreatic Tumor Models. Mol. Ther. Methods Clin. Dev. 2020, 17, 957–968. [Google Scholar] [CrossRef] [PubMed]
- Giannakopoulos, A.; Quiviger, M.; Stavrou, E.; Verras, M.; Marie, C.; Scherman, D.; Athanassiadou, A. Efficient Episomal Gene Transfer to Human Hepatic Cells Using the PFAR4-S/MAR Vector. Mol. Biol. Rep. 2019, 46, 3203–3211. [Google Scholar] [CrossRef] [PubMed]
- Pastor, M.; Johnen, S.; Harmening, N.; Quiviger, M.; Pailloux, J.; Kropp, M.; Walter, P.; Ivics, Z.; Izsvák, Z.; Thumann, G.; et al. The Antibiotic-Free PFAR4 Vector Paired with the Sleeping Beauty Transposon System Mediates Efficient Transgene Delivery in Human Cells. Mol.Ther. Nucleic Acids 2018, 11, 57–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turchiano, G.; Latella, M.C.; Gogol-Döring, A.; Cattoglio, C.; Mavilio, F.; Izsvák, Z.; Ivics, Z.; Recchia, A. Genomic Analysis of Sleeping Beauty Transposon Integration in Human Somatic Cells. PLoS ONE 2014, 9, e112712. [Google Scholar] [CrossRef] [PubMed]
- Boulikas, T. Homeotic Protein Binding Sites, Origins of Replication, and Nuclear Matrix Anchorage Sites Share the ATTA and ATTTA Motifs. J. Cell Biochem. 1992, 50, 111–123. [Google Scholar] [CrossRef] [PubMed]
- Ripin, N.; Boudet, J.; Duszczyk, M.M.; Hinniger, A.; Faller, M.; Krepl, M.; Gadi, A.; Schneider, R.J.; Šponer, J.; Meisner-Kober, N.C.; et al. Molecular Basis for AU-Rich Element Recognition and Dimerization by the HuR C-Terminal RRM. Proc. Natl. Acad. Sci. USA 2019, 116, 2935–2944. [Google Scholar] [CrossRef] [Green Version]
- Lieberman-Aiden, E.; van Berkum, N.L.; Williams, L.; Imakaev, M.; Ragoczy, T.; Telling, A.; Amit, I.; Lajoie, B.R.; Sabo, P.J.; Dorschner, M.O.; et al. Comprehensive Mapping of Long-Range Interactions Reveals Folding Principles of the Human Genome. Science 2009, 326, 289–293. [Google Scholar] [CrossRef] [Green Version]
- Dixon, J.R.; Selvaraj, S.; Yue, F.; Kim, A.; Li, Y.; Shen, Y.; Hu, M.; Liu, J.S.; Ren, B. Topological Domains in Mammalian Genomes Identified by Analysis of Chromatin Interactions. Nature 2012, 485, 376–380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zirkel, A.; Papantonis, A. Transcription as a Force Partitioning the Eukaryotic Genome. Biol. Chem. 2014, 395, 1301–1305. [Google Scholar] [CrossRef] [PubMed]
- Van Steensel, B.; Furlong, E.E.M. The Role of Transcription in Shaping the Spatial Organization of the Genome. Nat. Rev. Mol. Cell Biol. 2019, 20, 327–337. [Google Scholar] [CrossRef] [PubMed]
- Karmodiya, K.; Krebs, A.R.; Oulad-Abdelghani, M.; Kimura, H.; Tora, L. H3K9 and H3K14 Acetylation Co-Occur at Many Gene Regulatory Elements, While H3K14ac Marks a Subset of Inactive Inducible Promoters in Mouse Embryonic Stem Cells. BMC Genom. 2012, 13, 424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santos-Rosa, H.; Schneider, R.; Bannister, A.J.; Sherriff, J.; Bernstein, B.E.; Emre, N.C.; Schreiber, S.L.; Mellor, J.; Kouzarides, T. Active Genes Are Tri-Methylated at K4 of Histone H3. Nature 2002, 419, 407–411. [Google Scholar] [CrossRef] [PubMed]
- Koch, C.M.; Andrews, R.M.; Flicek, P.; Dillon, S.C.; Karaoz, U.; Clelland, G.K.; Wilcox, S.; Beare, D.M.; Fowler, J.C.; Couttet, P.; et al. The Landscape of Histone Modifications across 1% of the Human Genome in Five Human Cell Lines. Genome Res. 2007, 17, 691–707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rupprecht, S.; Lipps, H.J. Cell Cycle Dependent Histone Dynamics of an Episomal Non-Viral Vector. Gene 2009, 439, 95–101. [Google Scholar] [CrossRef] [PubMed]
- Deutsch, M.J.; Ott, E.; Papior, P.; Schepers, A. The Latent Origin of Replication of Epstein-Barr Virus Directs Viral Genomes to Active Regions of the Nucleus. J. Virol. 2009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hodin, T.L.; Najrana, T.; Yates, J.L. Efficient Replication of Epstein-Barr Virus-Derived Plasmids Requires Tethering by EBNA1 to Host Chromosomes. J. Virol. 2013, 87, 13020–13028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papantonis, A.; Kohro, T.; Baboo, S.; Larkin, J.D.; Deng, B.; Short, P.; Tsutsumi, S.; Taylor, S.; Kanki, Y.; Kobayashi, M.; et al. TNFalpha Signals through Specialized Factories Where Responsive Coding and MiRNA Genes Are Transcribed. EMBO J. 2012, 31, 4404–4414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, M.; Cook, P.R. Similar Active Genes Cluster in Specialized Transcription Factories. J. Cell Biol. 2008, 181, 615–623. [Google Scholar] [CrossRef] [Green Version]
- Papantonis, A.; Cook, P.R. Fixing the Model for Transcription: The DNA Moves, Not the Polymerase. Transcription 2011, 2, 41–44. [Google Scholar] [CrossRef]
- Gogol-Döring, A.; Ammar, I.; Gupta, S.; Bunse, M.; Miskey, C.; Chen, W.; Uckert, W.; Schulz, T.F.; Izsvák, Z.; Ivics, Z. Genome-Wide Profiling Reveals Remarkable Parallels Between Insertion Site Selection Properties of the MLV Retrovirus and the PiggyBac Transposon in Primary Human CD4(+) T Cells. Mol. Ther. 2016, 24, 592–606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zayed, H.; Izsvák, Z.; Walisko, O.; Ivics, Z. Development of Hyperactive Sleeping Beauty Transposon Vectors by Mutational Analysis. Mol. Ther. 2004, 9, 292–304. [Google Scholar] [CrossRef] [PubMed]
- Moldt, B.; Miskey, C.; Staunstrup, N.H.; Gogol-Döring, A.; Bak, R.O.; Sharma, N.; Mátés, L.; Izsvák, Z.; Chen, W.; Ivics, Z.; et al. Comparative Genomic Integration Profiling of Sleeping Beauty Transposons Mobilized with High Efficacy from Integrase-Defective Lentiviral Vectors in Primary Human Cells. Mol. Ther. 2011, 19, 1499–1510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Staunstrup, N.H.; Moldt, B.; Mátés, L.; Villesen, P.; Jakobsen, M.; Ivics, Z.; Izsvák, Z.; Mikkelsen, J.G. Hybrid Lentivirus-Transposon Vectors with a Random Integration Profile in Human Cells. Mol. Ther. 2009, 17, 1205–1214. [Google Scholar] [CrossRef] [PubMed]
- Vink, C.A.; Gaspar, H.B.; Gabriel, R.; Schmidt, M.; McIvor, R.S.; Thrasher, A.J.; Qasim, W. Sleeping Beauty Transposition from Nonintegrating Lentivirus. Mol. Ther. 2009, 17, 1197–1204. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Muck-Hausl, M.; Wang, J.; Sun, C.; Gebbing, M.; Miskey, C.; Ivics, Z.; Izsvak, Z.; Ehrhardt, A. Integration Profile and Safety of an Adenovirus Hybrid-Vector Utilizing Hyperactive Sleeping Beauty Transposase for Somatic Integration. PLoS ONE 2013, 8, e75344. [Google Scholar] [CrossRef] [Green Version]
- Bestor, T.H. Gene Silencing as a Threat to the Success of Gene Therapy. J. Clin. Investig. 2000, 105, 409–411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stein, S.; Ott, M.G.; Schultze-Strasser, S.; Jauch, A.; Burwinkel, B.; Kinner, A.; Schmidt, M.; Krämer, A.; Schwäble, J.; Glimm, H.; et al. Genomic Instability and Myelodysplasia with Monosomy 7 Consequent to EVI1 Activation after Gene Therapy for Chronic Granulomatous Disease. Nat. Med. 2010, 16, 198–204. [Google Scholar] [CrossRef] [Green Version]
- Cavazzana-Calvo, M.; Payen, E.; Negre, O.; Wang, G.; Hehir, K.; Fusil, F.; Down, J.; Denaro, M.; Brady, T.; Westerman, K.; et al. Transfusion Independence and HMGA2 Activation after Gene Therapy of Human β-Thalassaemia. Nature 2010, 467, 318–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yant, S.R.; Huang, Y.; Akache, B.; Kay, M.A. Site-Directed Transposon Integration in Human Cells. Nucleic Acids Res. 2007, 35, e50. [Google Scholar] [CrossRef] [PubMed]
- Voigt, K.; Gogol-Döring, A.; Miskey, C.; Chen, W.; Cathomen, T.; Izsvák, Z.; Ivics, Z. Retargeting Sleeping Beauty Transposon Insertions by Engineered Zinc Finger DNA-Binding Domains. Mol. Ther. 2012, 20, 1852–1862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ammar, I.; Gogol-Döring, A.; Miskey, C.; Chen, W.; Cathomen, T.; Izsvák, Z.; Ivics, Z. Retargeting Transposon Insertions by the Adeno-Associated Virus Rep Protein. Nucleic Acids Res. 2012, 40, 6693–6712. [Google Scholar] [CrossRef] [Green Version]
- Kovač, A.; Miskey, C.; Menzel, M.; Grueso, E.; Gogol-Döring, A.; Ivics, Z. RNA-Guided Retargeting of Sleeping Beauty Transposition in Human Cells. Elife 2020, 9, e53868. [Google Scholar] [CrossRef] [PubMed]
- Kovač, A.; Ivics, Z. Specifically Integrating Vectors for Targeted Gene Delivery: Progress and Prospects. Cell Gene Ther. Insights 2017, 3, 103–123. [Google Scholar] [CrossRef]
- Waehler, R.; Russell, S.J.; Curiel, D.T. Engineering Targeted Viral Vectors for Gene Therapy. Nat. Rev. Genet. 2007, 8, 573–587. [Google Scholar] [CrossRef] [PubMed]
- Kreppel, F.; Hagedorn, C. Capsid and Genome Modification Strategies to Reduce the Immunogenicity of Adenoviral Vectors. Int. J. Mol. Sci. 2021, 22, 2417. [Google Scholar] [CrossRef] [PubMed]
- Kreppel, F.; Kochanek, S. Modification of Adenovirus Gene Transfer Vectors With Synthetic Polymers: A Scientific Review and Technical Guide. Mol. Ther. 2008, 16, 16–29. [Google Scholar] [CrossRef]
- Baden, L.R.; El Sahly, H.M.; Essink, B.; Kotloff, K.; Frey, S.; Novak, R.; Diemert, D.; Spector, S.A.; Rouphael, N.; Creech, C.B.; et al. Efficacy and Safety of the MRNA-1273 SARS-CoV-2 Vaccine. N. Engl. J. Med. 2021, 384, 403–416. [Google Scholar] [CrossRef] [PubMed]
- Thomas, S.J.; Moreira, E.D.; Kitchin, N.; Absalon, J.; Gurtman, A.; Lockhart, S.; Perez, J.L.; Pérez Marc, G.; Polack, F.P.; Zerbini, C.; et al. Safety and Efficacy of the BNT162b2 MRNA Covid-19 Vaccine through 6 Months. N. Engl. J. Med. 2021, 385, 1761–1773. [Google Scholar] [CrossRef]
- Hou, X.; Zaks, T.; Langer, R.; Dong, Y. Lipid Nanoparticles for MRNA Delivery. Nat. Rev. Mater. 2021, 6, 1078–1094. [Google Scholar] [CrossRef] [PubMed]
- Steffens, R.C.; Wagner, E. Directing the Way—Receptor and Chemical Targeting Strategies for Nucleic Acid Delivery. Pharm. Res. 2022, 1–30. [Google Scholar] [CrossRef] [PubMed]
- Amberger, M.; Ivics, Z. Latest Advances for the Sleeping Beauty Transposon System: 23 Years of Insomnia but Prettier than Ever: Refinement and Recent Innovations of the Sleeping Beauty Transposon System Enabling Novel, Nonviral Genetic Engineering Applications. Bioessays 2020, 42, e2000136. [Google Scholar] [CrossRef] [PubMed]
- Dorigo, O.; Gil, J.S.; Gallaher, S.D.; Tan, B.T.; Castro, M.G.; Lowenstein, P.R.; Calos, M.P.; Berk, A.J. Development of a Novel Helper-Dependent Adenovirus-Epstein-Barr Virus Hybrid System for the Stable Transformation of Mammalian Cells. J. Virol. 2004, 78, 6556–6566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kreppel, F.; Kochanek, S. Long-Term Transgene Expression in Proliferating Cells Mediated by Episomally Maintained High-Capacity Adenovirus Vectors. J. Virol. 2004, 78, 9–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gil, J.S.; Gallaher, S.D.; Berk, A.J. Delivery of an EBV Episome by a Self-Circularizing Helper-Dependent Adenovirus: Long-Term Transgene Expression in Immunocompetent Mice. Gene Ther. 2010, 17, 1288–1293. [Google Scholar] [CrossRef] [PubMed]
- Hagedorn, C.; Schnödt-Fuchs, M.; Boehme, P.; Abdelrazik, H.; Lipps, H.J.; Büning, H. S/MAR Element Facilitates Episomal Long-Term Persistence of Adeno-Associated Virus Vector Genomes in Proliferating Cells. Hum. Gene Ther. 2017, 28, 1169–1179. [Google Scholar] [CrossRef] [PubMed]
- Turunen, T.A.K.; Laakkonen, J.P.; Alasaarela, L.; Airenne, K.J.; Ylä-Herttuala, S. Sleeping Beauty-Baculovirus Hybrid Vectors for Long-Term Gene Expression in the Eye. J. Gene Med. 2014, 16, 40–53. [Google Scholar] [CrossRef] [PubMed]
- Hausl, M.A.; Zhang, W.; Müther, N.; Rauschhuber, C.; Franck, H.G.; Merricks, E.P.; Nichols, T.C.; Kay, M.A.; Ehrhardt, A. Hyperactive Sleeping Beauty Transposase Enables Persistent Phenotypic Correction in Mice and a Canine Model for Hemophilia B. Mol. Ther. 2010, 18, 1896–1906. [Google Scholar] [CrossRef] [PubMed]
- Voigtlander, R.; Haase, R.; Muck-Hausl, M.; Zhang, W.; Boehme, P.; Lipps, H.J.; Schulz, E.; Baiker, A.; Ehrhardt, A. A Novel Adenoviral Hybrid-Vector System Carrying a Plasmid Replicon for Safe and Efficient Cell and Gene Therapeutic Applications. Mol. Ther. Nucleic Acids 2013, 2, e83. [Google Scholar] [CrossRef]
- Harraghy, N.; Gaussin, A.; Mermod, N. Sustained Transgene Expression Using MAR Elements. Curr. Gene Ther. 2008, 8, 353–366. [Google Scholar] [CrossRef] [PubMed]
- Grandjean, M.; Girod, P.-A.; Calabrese, D.; Kostyrko, K.; Wicht, M.; Yerly, F.; Mazza, C.; Beckmann, J.S.; Martinet, D.; Mermod, N. High-Level Transgene Expression by Homologous Recombination-Mediated Gene Transfer. Nucleic Acids Res. 2011, 39, e104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, W.; Hagedorn, C.; Schulz, E.; Lipps, H.-J.; Ehrhardt, A. Viral Hybrid-Vectors for Delivery of Autonomous Replicons. Curr. Gene Ther. 2014, 14, 10–23. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Kamihira, M. Development of Hybrid Viral Vectors for Gene Therapy. Biotechnol. Adv. 2013, 31, 208–223. [Google Scholar] [CrossRef] [PubMed]
- Raper, S.E.; Chirmule, N.; Lee, F.S.; Wivel, N.A.; Bagg, A.; Gao, G.; Wilson, J.M.; Batshaw, M.L. Fatal Systemic Inflammatory Response Syndrome in a Ornithine Transcarbamylase Deficient Patient Following Adenoviral Gene Transfer. Mol. Genet. Metab. 2003, 80, 148–158. [Google Scholar] [CrossRef] [PubMed]
- Zolgensma Acute Liver Failure Update. Available online: https://www.novartis.com/news/zolgensma-acute-liver-failure-update (accessed on 17 August 2022).
- Kreppel, F.; Gackowski, J.; Schmidt, E.; Kochanek, S. Combined Genetic and Chemical Capsid Modifications Enable Flexible and Efficient De- and Retargeting of Adenovirus Vectors. Mol. Ther. 2005, 12, 107–117. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| VECTOR SYSTEM | ADVANTAGES | DISADVANTAGES |
|---|---|---|
| EBV-based replicons | - stable transgene expression - high cloning capacity (>100 kb) - episomal maintenance - efficient modification of target cells - easy engineering, low-cost production | - contains viral elements - low delivery efficiencies (in vitro and in vivo) - limited targeting options - EBNA-1-related toxicity |
| Sleeping Beauty | - stable transgene expression - comprehensively studied safety profile - efficient modification of target cells | - risk of insertional mutagenesis - requires co-delivery of transposases - low delivery efficiencies (in vitro and in vivo) |
| S/MAR-based replicons | - lack of viral elements - episomal maintenance - easy engineering, low-cost production - low immunogenicity | - low establishment efficiencies - limited cloning capacity - low delivery efficiencies (in vitro and in vivo) - limited targeting options |
| Artificial Chromosomes | - unlimited cloning capacity - copy number control - nonintegrating - nonessential, additional chromosome | - complex construction - low delivery efficiencies (in vitro and in vivo) - construct-dependent inefficiencies in segregation to daughter cells possible - potential risks imposed by homologous recombination need to be analysed in depth |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kreppel, F.; Hagedorn, C. Episomes and Transposases—Utilities to Maintain Transgene Expression from Nonviral Vectors. Genes 2022, 13, 1872. https://doi.org/10.3390/genes13101872
Kreppel F, Hagedorn C. Episomes and Transposases—Utilities to Maintain Transgene Expression from Nonviral Vectors. Genes. 2022; 13(10):1872. https://doi.org/10.3390/genes13101872
Chicago/Turabian StyleKreppel, Florian, and Claudia Hagedorn. 2022. "Episomes and Transposases—Utilities to Maintain Transgene Expression from Nonviral Vectors" Genes 13, no. 10: 1872. https://doi.org/10.3390/genes13101872