
A Comprehensive Analysis of KRT19 Combined with Immune Infiltration to Predict Breast Cancer Prognosis


Abstract
:1. Introduction
2. Materials and Methods
2.1. Gene Expression and Clinicopathological Character Analysis
2.2. The Genetic Alterations Analysis
2.3. Enrichment Analysis of KRT19-Related Genes
2.4. Single-Cell Analysis
2.5. Immunity Analysis
2.6. Construction and Evaluation of Nomogram
3. Results
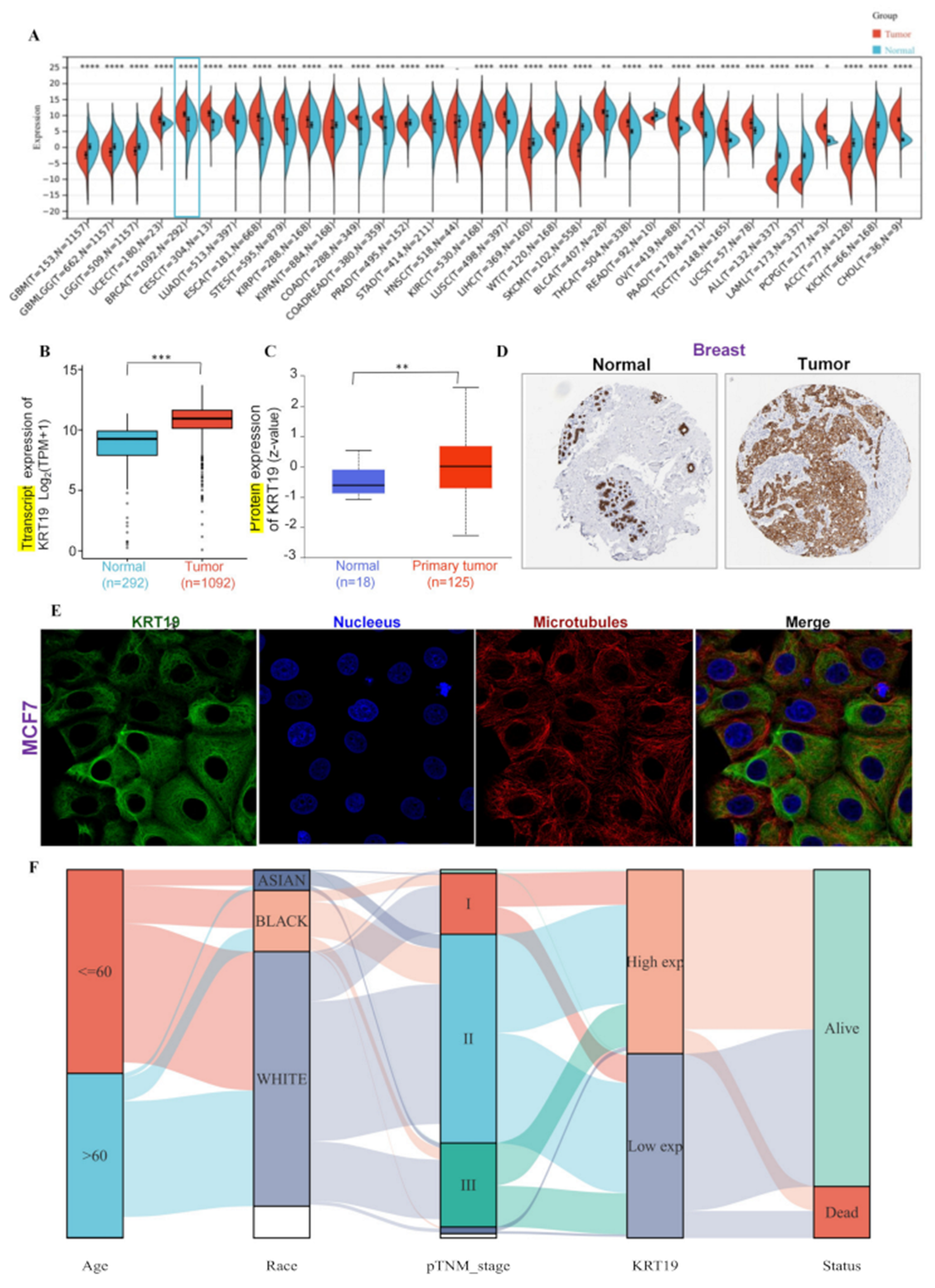
3.1. Expression of the KRT19 in BRCA and Clinicopathological Characteristics in BRCA Patients
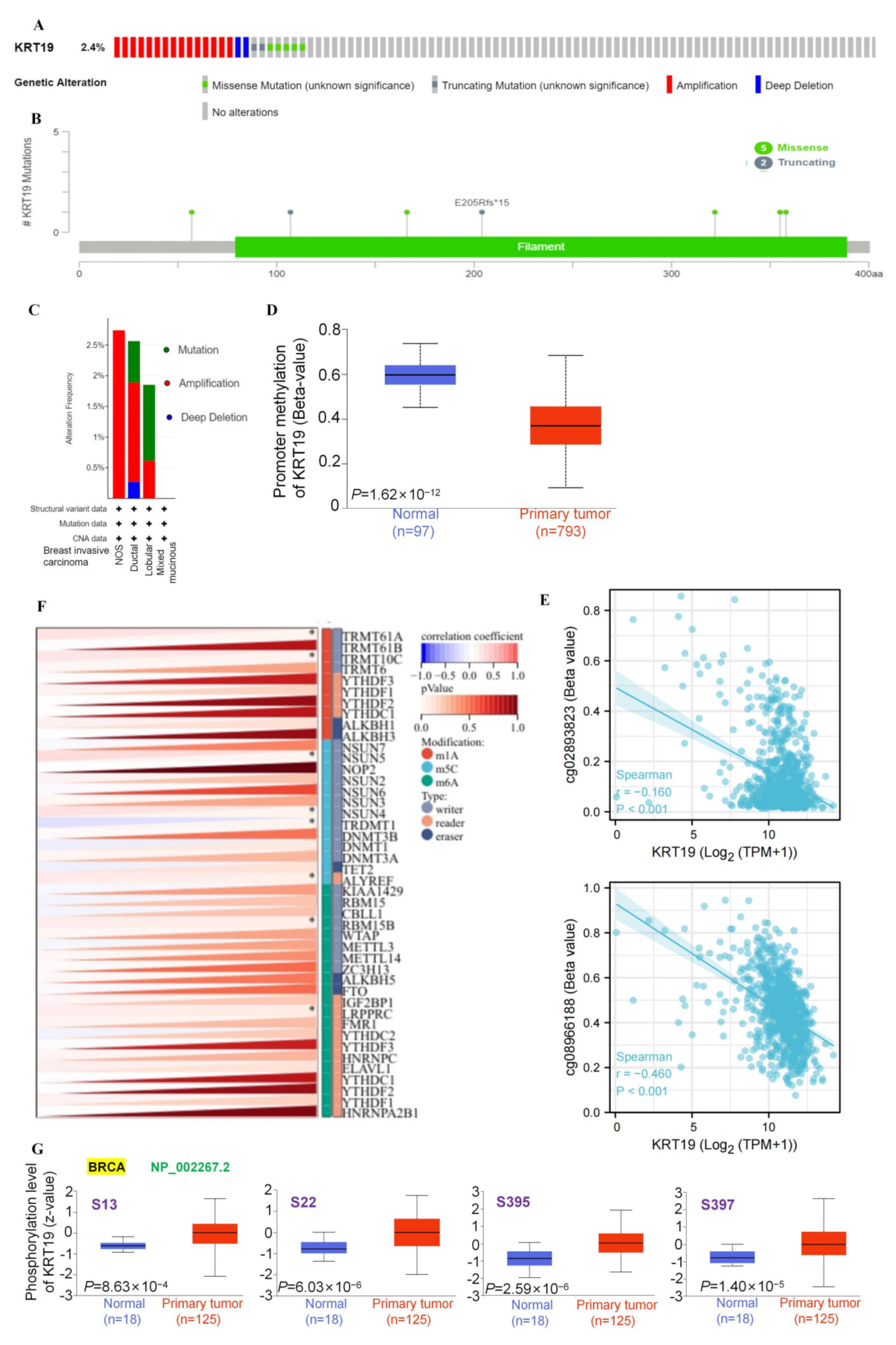
3.2. Epigenetic Variations and Genomic Heterogeneity of KRT19 in BRCA
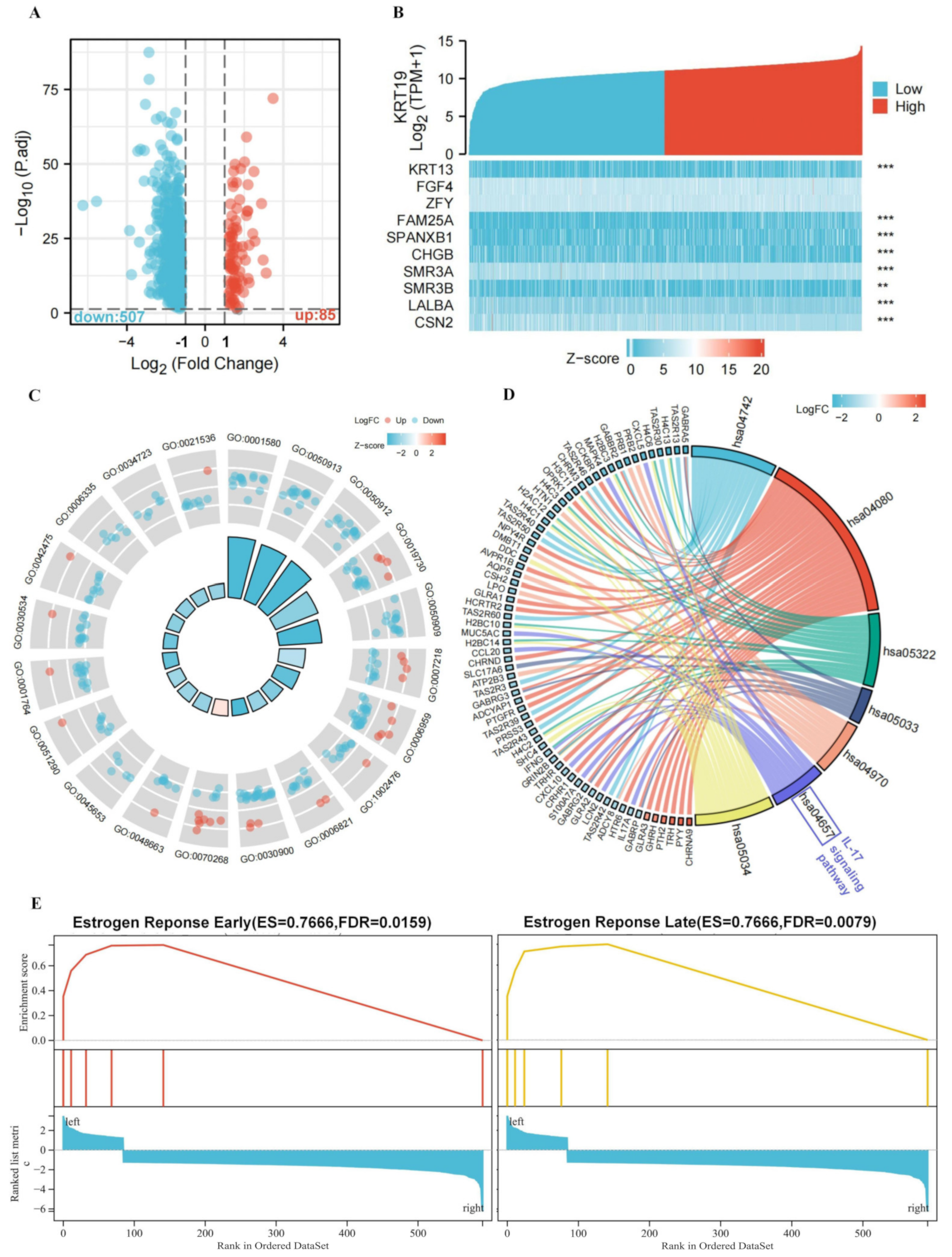
3.3. Functional Annotation of KRT19-Associated DEGs in BRCA
3.4. Biological Functions of the KRT19 in BRCA
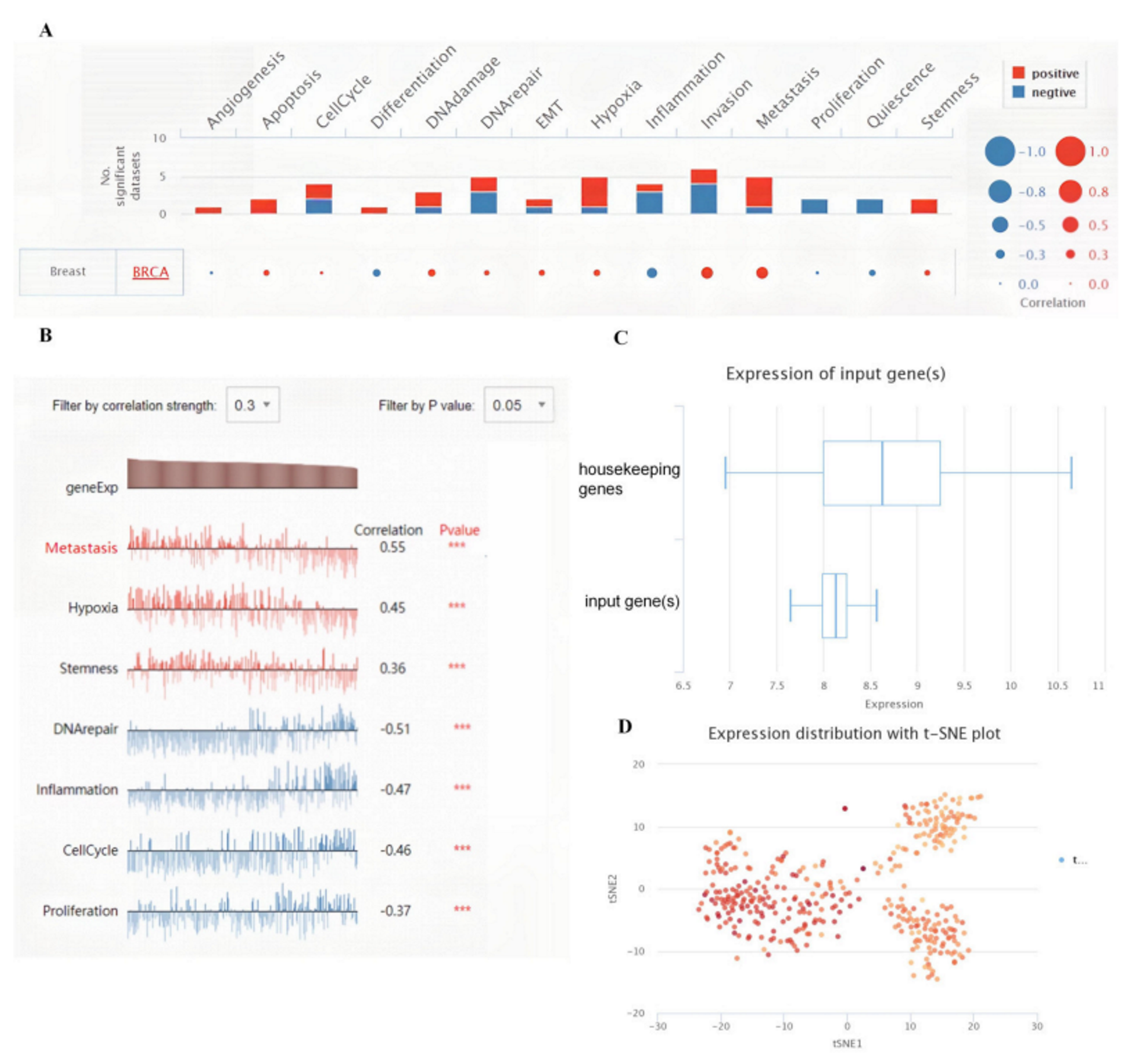
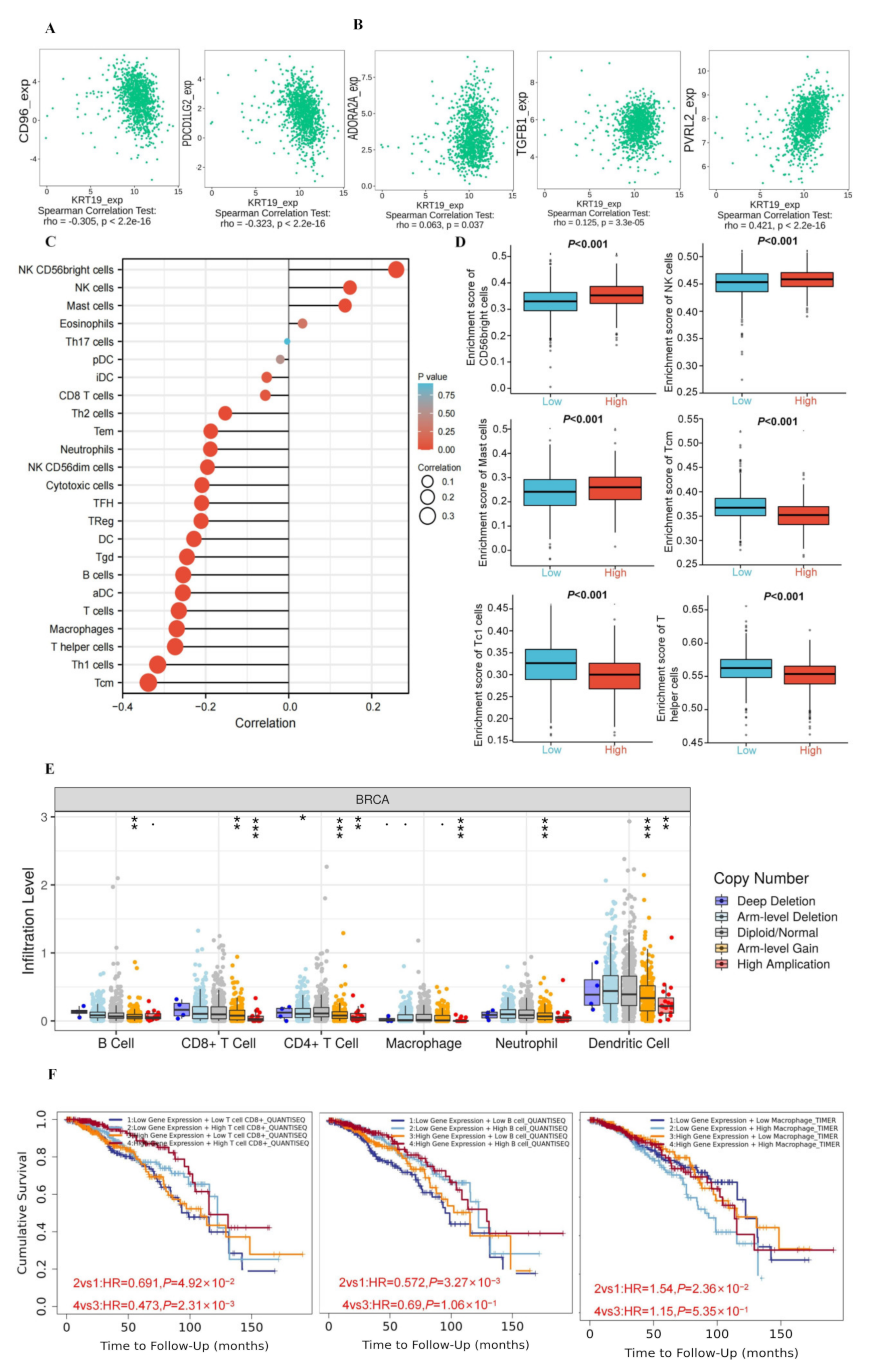
3.5. The Correlations between KRT19 Expression and Immunity in BRCA
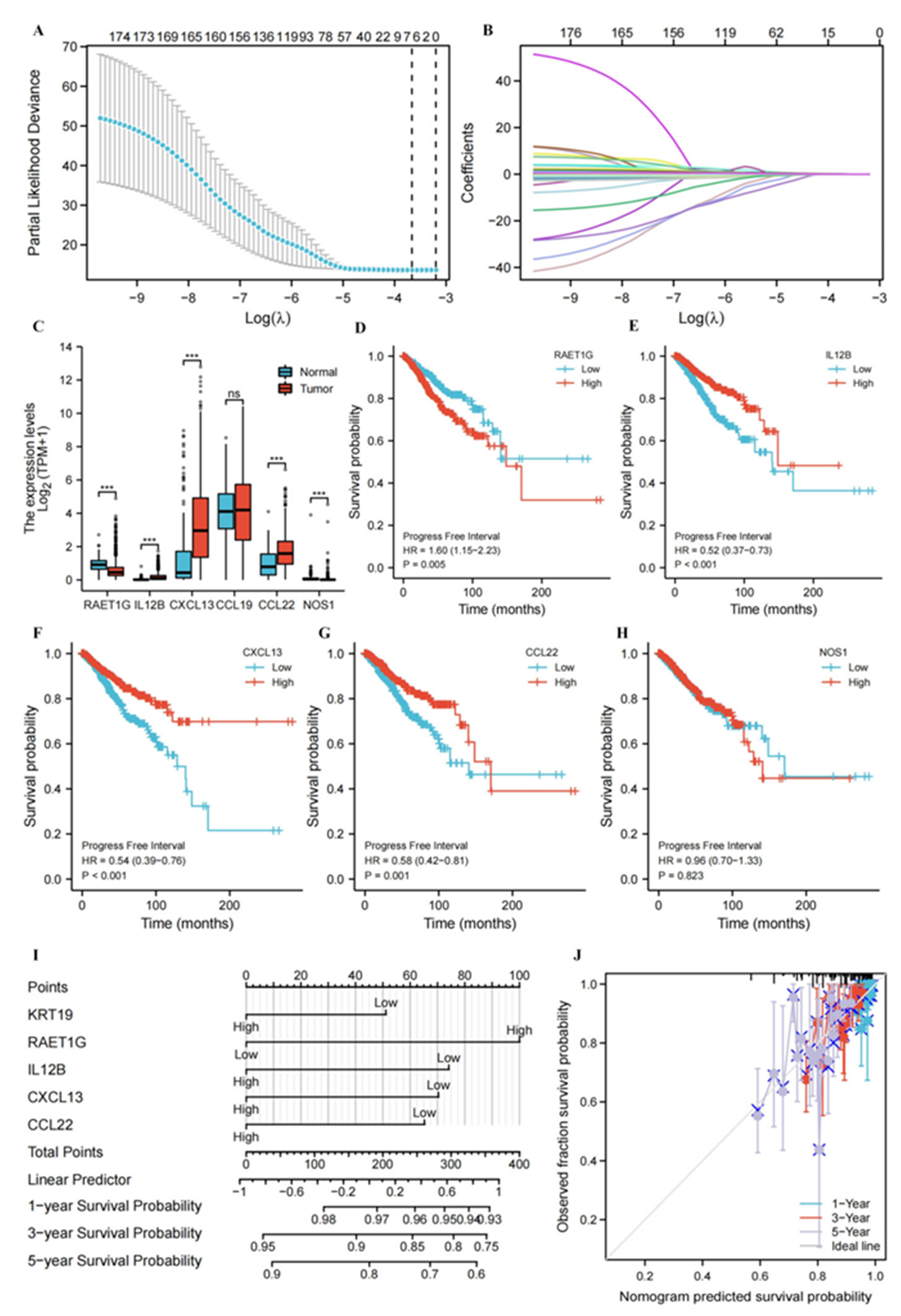
3.6. Construction and Evaluation of Nomogram Based on Genes Associated with the Cancer-immunity Cycle Signatures
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2022. CA A Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef]
- Lamy, P.-J.; Fina, F.; Bascoul-Mollevi, C.; Laberenne, A.-C.; Martin, P.-M.; Ouafik, L.; Jacot, W. Quantification and clinical relevance of gene amplification at chromosome 17q12-q21 in human epidermal growth factor receptor 2-amplified breast cancers. Breast Cancer Res. 2011, 13, R15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, J.S.; Han, K.H.; Kim, E.-K.; Moon, H.J.; Yoon, J.H.; Kim, M.J. Fine-Needle Aspirate CYFRA 21-1, an Innovative New Marker for Diagnosis of Axillary Lymph Node Metastasis in Breast Cancer Patients. Medicine 2015, 94, e811. [Google Scholar] [CrossRef] [PubMed]
- Nicolini, A.; Ferrari, P.; Rossi, G. Mucins and Cytokeratins as Serum Tumor Markers in Breast Cancer. Adv. Exp. Med. Biol. 2015, 867, 197–225. [Google Scholar] [CrossRef] [PubMed]
- Jochems, C.; Schlom, J. Tumor-infiltrating immune cells and prognosis: The potential link between conventional cancer therapy and immunity. Exp. Biol. Med. 2011, 236, 567–579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dieci, M.; Miglietta, F.; Guarneri, V. Immune Infiltrates in Breast Cancer: Recent Updates and Clinical Implications. Cells 2021, 10, 223. [Google Scholar] [CrossRef] [PubMed]
- Chin, L.; Andersen, J.; Futreal, P.A. Cancer genomics: From discovery science to personalized medicine. Nat. Med. 2011, 17, 297–303. [Google Scholar] [CrossRef]
- Lonsdale, J.; Thomas, J.; Salvatore, M.; Phillips, R.; Lo, E.; Shad, S.; Hasz, R.; Walters, G.; Garcia, F.; Young, N.; et al. GTEx Consortium. The Genotype-Tissue Expression (GTEx) project. Nat. Genet. 2013, 45, 580–585. [Google Scholar] [CrossRef]
- Chandrashekar, D.S.; Bashel, B.; Balasubramanya, S.A.H.; Creighton, C.J.; Ponce-Rodriguez, I.; Chakravarthi, B.V.S.K.; Varambally, S. UALCAN: A portal for facilitating tumor subgroup gene expression and survival analyses. Neoplasia 2017, 19, 649–658. [Google Scholar] [CrossRef]
- Chen, F.; Chandrashekar, D.S.; Varambally, S.; Creighton, C.J. Pan-cancer molecular subtypes revealed by mass-spectrometry-based proteomic characterization of more than 500 human cancers. Nat. Commun. 2019, 10, 5679. [Google Scholar] [CrossRef]
- Uhlén, M.; Zhang, C.; Lee, S.; Sjöstedt, E.; Fagerberg, L.; Bidkhori, G.; Benfeitas, R.; Arif, M.; Liu, Z.; Edfors, F.; et al. A pathology atlas of the human cancer transcriptome. Science 2017, 357, 2507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.E.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative Analysis of Complex Cancer Genomics and Clinical Profiles Using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koch, A.; De Meyer, T.; Jeschke, J.; Van Criekinge, W. MEXPRESS: Visualizing expression, DNA methylation and clinical TCGA data. BMC Genom. 2015, 16, 636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, W.; Song, Z.; Zhong, X.; Huang, M.; Shen, D.; Gao, P.; Qian, X.; Wang, M.; He, X.; Wang, T.; et al. Sangerbox: A comprehensive, interaction-friendly clinical bioinformatics analysis platform. iMeta 2022, 1, e36. [Google Scholar] [CrossRef]
- Yuan, H.; Yan, M.; Zhang, G.; Liu, W.; Deng, C.; Liao, G.; Xu, L.; Luo, T.; Yan, H.; Long, Z.; et al. CancerSEA: A cancer single-cell state atlas. Nucleic Acids Res. 2018, 47, D900–D908. [Google Scholar] [CrossRef] [Green Version]
- Ru, B.; Wong, C.N.; Tong, Y.; Zhong, J.Y.; Zhong, S.S.W.; Wu, W.C.; Chu, K.C.; Wong, C.Y.; Lau, C.Y.; Chen, I.; et al. TISIDB: An integrated repository portal for tumor–immune system interactions. Bioinformatics 2019, 35, 4200–4202. [Google Scholar] [CrossRef]
- Li, T.; Fu, J.; Zeng, Z.; Cohen, D.; Li, J.; Chen, Q.; Li, B.; Liu, X.S. TIMER2.0 for analysis of tumor-infiltrating immune cells. Nucleic Acids Res. 2020, 48, W509–W514. [Google Scholar] [CrossRef]
- Xu, L.; Deng, C.; Pang, B.; Zhang, X.; Liu, W.; Liao, G.; Yuan, H.; Cheng, P.; Li, F.; Long, Z.; et al. TIP: A Web Server for Resolving Tumor Immunophenotype Profiling. Cancer Res. 2018, 78, 6575–6580. [Google Scholar] [CrossRef] [Green Version]
- Stuart, T.; Satija, R. Integrative single-cell analysis. Nat. Rev. Genet. 2019, 20, 257–272. [Google Scholar] [CrossRef]
- Kabir, N.N.; Rönnstrand, L.; Kazi, J.U. Keratin 19 expression correlates with poor prognosis in breast cancer. Mol. Biol. Rep. 2014, 41, 7729–7735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheikh, A.; Hussain, S.A.; Ghori, Q.; Naeem, N.; Fazil, A.; Giri, S.; Sathian, B.; Mainali, P.; Al Tamimi, D.M. The Spectrum of Genetic Mutations in Breast Cancer. Asian Pac. J. Cancer Prev. 2015, 16, 2177–2185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moore, L.D.; Le, T.; Fan, G. DNA methylation and its basic function. Neuropsychopharmacology 2013, 38, 23–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kulis, M.; Esteller, M. DNA methylation and cancer. Adv. Genet. 2010, 70, 27–56. [Google Scholar]
- De Almeida, B.P.; Apolónio, J.D.; Binnie, A.; Castelo-Branco, P. Roadmap of DNA methylation in breast cancer identifies novel prognostic biomarkers. BMC Cancer 2019, 19, 219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, V.; Ram, M.; Kumar, R.; Prasad, R.; Roy, B.K.; Singh, K.K. Phosphorylation: Implications in Cancer. J. Protein Chem. 2017, 36, 1–6. [Google Scholar] [CrossRef]
- Haruehanroengra, P.; Zheng, Y.Y.; Zhou, Y.; Huang, Y.; Sheng, J. RNA modifications and cancer. RNA Biol. 2020, 17, 1560–1575. [Google Scholar] [CrossRef]
- Li, X.; Bechara, R.; Zhao, J.; McGeachy, M.J.; Gaffen, S.L. IL-17 receptor–based signaling and implications for disease. Nat. Immunol. 2019, 20, 1594–1602. [Google Scholar] [CrossRef]
- Zhang, Q.; Liu, S.; Parajuli, K.R.; Zhang, W.; Zhang, K.; Mo, Z.; Liu, J.; Chen, Z.; Yang, S.; Wang, A.R.; et al. Interleukin-17 promotes prostate cancer via MMP7-induced epithelial-to-mesenchymal transition. Oncogene 2016, 36, 687–699. [Google Scholar] [CrossRef] [Green Version]
- Alinejad, V.; Dolati, S.; Motallebnezhad, M.; Yousefi, M. The role of IL17B-IL17RB signaling pathway in breast cancer. Biomed. Pharmacother. 2017, 88, 795–803. [Google Scholar] [CrossRef]
- Salmaninejad, A.; Valilou, S.F.; Shabgah, A.G.; Aslani, S.; Alimardani, M.; Pasdar, A.; Sahebkar, A. PD-1/PD-L1 pathway: Basic biology and role in cancer immunotherapy. J. Cell. Physiol. 2019, 234, 16824–16837. [Google Scholar] [CrossRef] [PubMed]
- Le Bouteiller, P.; Tabiasco, J.; Polgar, B.; Kozma, N.; Giustiniani, J.; Siewiera, J.; Berrebi, A.; Aguerre-Girr, M.; Bensussan, A.; Jabrane-Ferrat, N. CD160: A unique activating NK cell receptor. Immunol. Lett. 2011, 138, 93–96. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.S.; Mellman, I. Oncology Meets Immunology: The Cancer-Immunity Cycle. Immunity 2013, 39, 1–10. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Character | Total (N) | Univariate Analysis | Multivariate Analysis | ||
|---|---|---|---|---|---|
| Hazard Ratio (95% CI) | p Value | Hazard Ratio (95% CI) | p Value | ||
| Age | 1082 | ||||
| ≤60 | 601 | Reference | |||
| >60 | 481 | 1.253 (0.904–1.738) | 0.175 | ||
| Race | 993 | ||||
| Asian | 60 | Reference | |||
| White | 753 | 0.832 (0.338–2.049) | 0.689 | ||
| Black or African American | 180 | 0.947 (0.364–2.465) | 0.912 | ||
| T stage | 1079 | ||||
| T1 | 276 | Reference | |||
| T2 | 629 | 1.615 (1.042–2.501) | 0.032 | 1.342 (0.818–2.204) | 0.244 |
| T3 | 139 | 2.213 (1.290–3.798) | 0.004 | 1.211 (0.615–2.385) | 0.579 |
| T4 | 35 | 6.258 (3.262–12.008) | <0.001 | 2.775 (1.227–6.279) | 0.014 |
| N stage | 1063 | ||||
| N0 | 514 | Reference | |||
| N1 | 357 | 1.981 (1.331–2.948) | <0.001 | 1.481 (0.957–2.290) | 0.078 |
| N2 | 116 | 2.481 (1.441–4.272) | 0.001 | 2.154 (1.200–3.864) | 0.010 |
| N3 | 76 | 4.961 (2.833–8.688) | <0.001 | 2.436 (1.159–5.124) | 0.019 |
| M stage | 922 | ||||
| M0 | 902 | Reference | |||
| M1 | 20 | 8.315 (4.829–14.315) | <0.001 | 3.408 (1.699–6.838) | <0.001 |
| KRT19 | 1082 | ||||
| Low | 540 | Reference | |||
| High | 542 | 0.856 (0.617–1.187) | 0.350 | ||
| RAET1G | 1082 | ||||
| Low | 541 | Reference | |||
| High | 541 | 1.604 (1.152–2.234) | 0.005 | 1.912 (1.318–2.774) | <0.001 |
| IL12B | 1082 | ||||
| Low | 541 | Reference | |||
| High | 541 | 0.519 (0.371–0.726) | <0.001 | 0.809 (0.511–1.279) | 0.364 |
| CXCL13 | 1082 | ||||
| Low | 540 | Reference | |||
| High | 542 | 0.543 (0.389–0.758) | <0.001 | 0.756 (0.491–1.166) | 0.206 |
| CCL22 | 1082 | ||||
| Low | 540 | Reference | |||
| High | 542 | 0.584 (0.420–0.812) | 0.001 | 0.714 (0.472–1.079) | 0.110 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mi, L.; Liang, N.; Sun, H. A Comprehensive Analysis of KRT19 Combined with Immune Infiltration to Predict Breast Cancer Prognosis. Genes 2022, 13, 1838. https://doi.org/10.3390/genes13101838
Mi L, Liang N, Sun H. A Comprehensive Analysis of KRT19 Combined with Immune Infiltration to Predict Breast Cancer Prognosis. Genes. 2022; 13(10):1838. https://doi.org/10.3390/genes13101838
Chicago/Turabian StyleMi, Lusi, Nan Liang, and Hui Sun. 2022. "A Comprehensive Analysis of KRT19 Combined with Immune Infiltration to Predict Breast Cancer Prognosis" Genes 13, no. 10: 1838. https://doi.org/10.3390/genes13101838