
A Comparative Cross-Platform Analysis to Identify Potential Biomarker Genes for Evaluation of Teratozoospermia and Azoospermia
 , ,
, ,  ,
,  , , ,
, , ,  , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Microarray Data
2.2. DEG Screening and Meta-Analyses
2.3. Comparative Analyses
2.4. Protein-Protein Interaction (PPI) Network
2.5. Pathway Enrichment Analyses
3. Results
3.1. Expression of Up- and Down-Regulated Genes (i.e., DEGs)
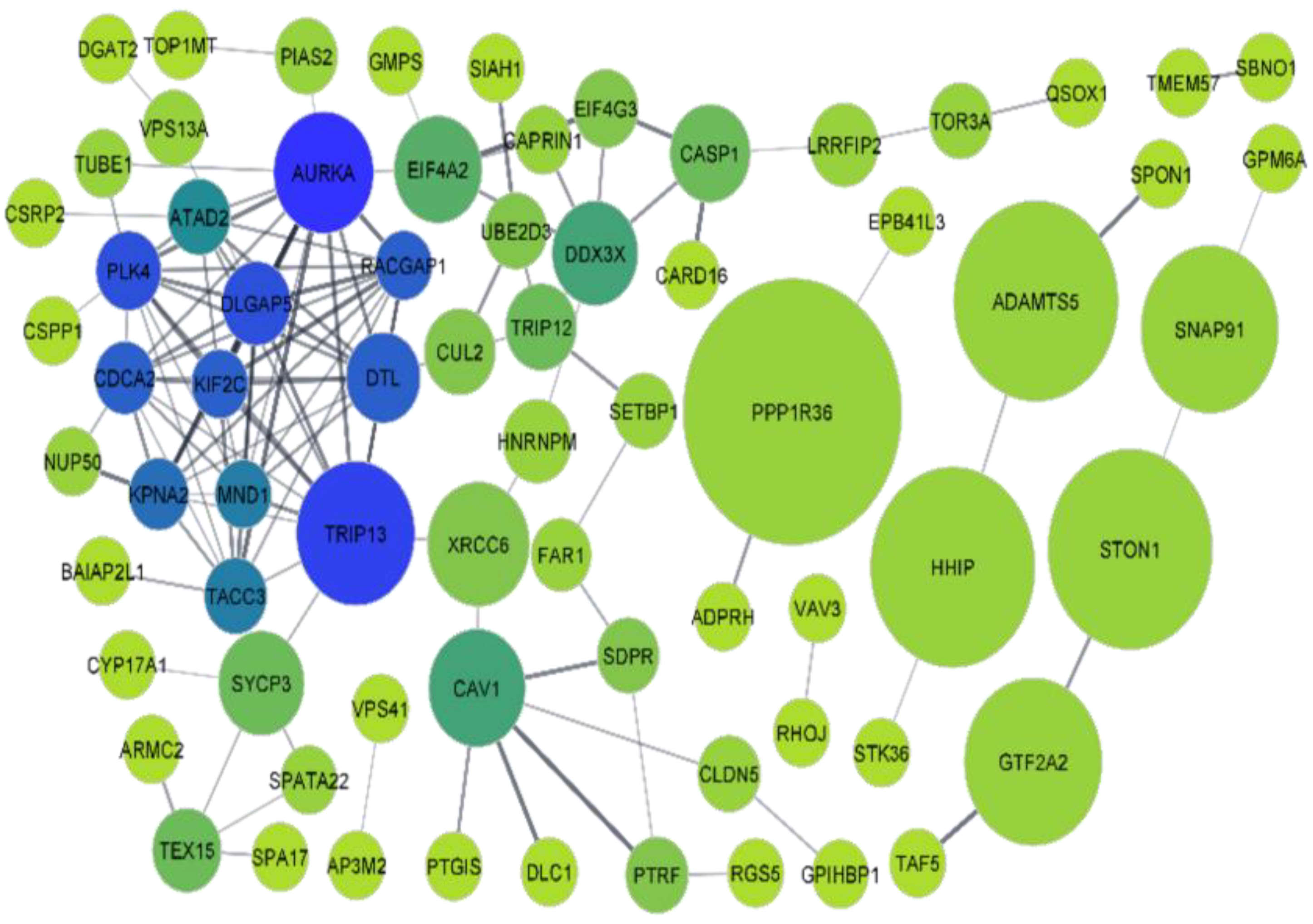
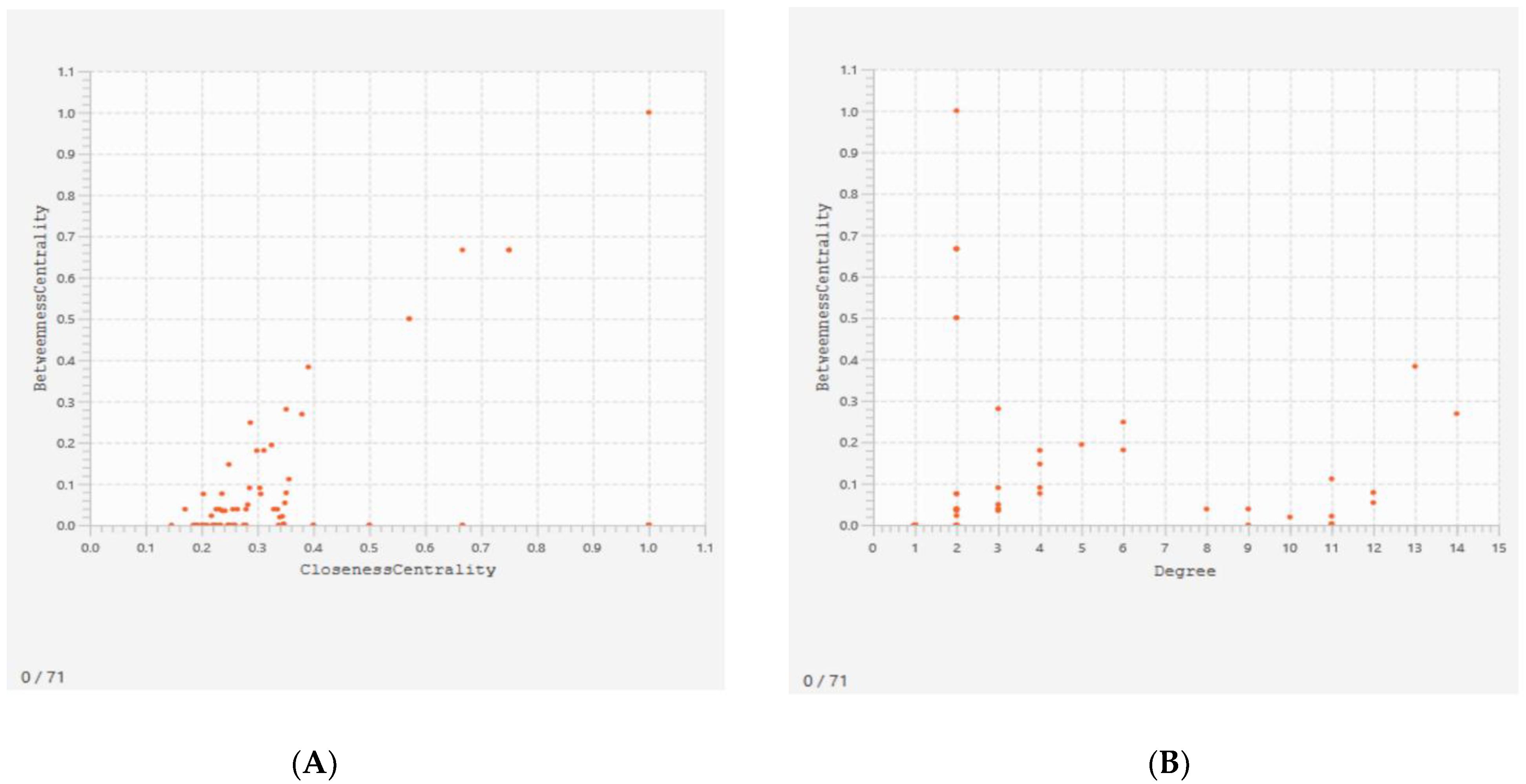
3.2. Protein-Protein Interaction (PPI) Network
3.3. Pathway Enrichment Analyses
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rhouma, B.; Okutman, O.; Muller, J.; Benkhalifa, M.; Bahri, H.; Rhouma, B.; Tebourbi, O.; Viville, S. Genetic aspects of male infertility: From bench to clinic. Gynecol. Obstet. Fertil. Senol. 2018, 47, 54–62. [Google Scholar] [PubMed]
- Vander Borght, M.; Wyns, C. Fertility and infertility: Definition and epidemiology. Clin. Biochem. 2018, 62, 2–10. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, A.; Mulgund, A.; Hamada, A.; Chyatte, M.R. A unique view on male infertility around the globe. Reproduct. Biol. Endocrinol. 2015, 13, 37. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, P.; Dutta, S.; Krajewska-Kulak, E. The disappearing sperms: Analysis of reports published between 1980 and 2015. Am. J. Mens Health 2017, 11, 1279–1304. [Google Scholar] [CrossRef] [PubMed]
- Dabaja, A.A.; Schlegel, P.N. Medical treatment of male infertility. Transl. Androl. Urol. 2014, 3, 9–16. [Google Scholar]
- Daneshmandpour, Y.; Bahmanpour, Z.; Hamzeiy, H.; Mazaheri Moghaddam, M.; Mazaheri Moghaddam, M.; Khademi, B.; Sakhinia, E. Micrornas association with azoospermia, oligospermia, asthenozoospermia, and teratozoospermia: A systematic review. J. Assist. Reprod. Genet. 2020, 37, 763–775. [Google Scholar] [CrossRef]
- Candela, L.; Boeri, L.; Capogrosso, P.; Cazzaniga, W.; Pozzi, E.; Belladelli, F.; Baudo, A.; Ravizzoli, A.; Ventimiglia, E.; Viganò, P. Correlation among isolated teratozoospermia, sperm DNA fragmentation and markers of systemic inflammation in primary infertile men. PLoS ONE 2021, 16, e0251608. [Google Scholar] [CrossRef]
- Mostafa Nayel, D.; Salah El Din Mahrous, H.; El Din Khalifa, E.; Kholeif, S.; Mohamed Elhady, G. The effect of teratozoospermia on sex chromosomes in human embryos. Appl. Clin. Genet. 2021, 14, 125–144. [Google Scholar] [CrossRef]
- Dziminski, M.A.; Roberts, J.D.; Simmons, L.W. Sperm morphology, motility and fertilisation capacity in the myobatrachid frog crinia georgiana. Reprod. Fertil. Dev. 2010, 22, 516–522. [Google Scholar] [CrossRef]
- Cocuzza, M.; Alvarenga, C.; Pagani, R. The epidemiology and etiology of azoospermia. Clinics 2013, 68 (Suppl. 1), 15–26. [Google Scholar] [CrossRef]
- Han, B.; Wang, L.; Yu, S.; Ge, W.; Li, Y.; Jiang, H.; Shen, W.; Sun, Z. One potential biomarker for teratozoospermia identified by in-depth integrative analysis of multiple microarray data. Aging 2021, 13, 10208–10224. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, P.; Dutta, S.; Karkada, I.R.; Chinni, S.V. Endocrinopathies and male infertility. Life 2022, 12, 10. [Google Scholar] [CrossRef] [PubMed]
- Kumar, N.; Singh, A.K. Trends of male factor infertility, an important cause of infertility: A review of literature. J. Hum. Reprod. Sci 2015, 8, 191–196. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Swefloff, R.S. Limitations of semen analysis as a test of male fertility and anticipated needs from newer tests. Fertil. Steril. 2014, 102, 1502–1507. [Google Scholar] [CrossRef] [PubMed]
- Boissonnas, C.C.; Jouannet, P.; Jammes, H. Epigenetic disorders and male subfertility. Fertil. Steril. 2013, 99, 624–631. [Google Scholar] [CrossRef] [PubMed]
- Gannon, J.R.; Emery, B.R.; Jenkins, T.G.; Carell, D.T. The sperm epigenome: Implications for the embryo. Adv. Exp. Med. Biol. 2014, 791, 53–66. [Google Scholar] [PubMed]
- Klaver, R.; Gromoll, J. Bringing eoigenetics into the diagnostics of the andrology laboratory: Challenges and perspectives. Asian J. Androl. 2014, 16, 669–674. [Google Scholar]
- de Mateo, S.; Sassone Corsi, P. Regulation of spermatogenesis by small non-coding RNAs: Role of the germ granule. Semin. Cell Dev. Biol. 2014, 29, 84–92. [Google Scholar] [CrossRef]
- Jodar, M.; Elvaraju, S.; Sendler, E.; Diamond, M.P.; Krawetz, S.A. The presence, role and clinical use of spermatozonal RNAs. Hum. Reprod. Update 2013, 19, 604–624. [Google Scholar] [CrossRef]
- Hotaling, J.; Carell, D.T. Clinical genetic testing for male factor infertility: Current applications and future directions. Andrology 2014, 2, 339–350. [Google Scholar] [CrossRef]
- Li, C.; Zhou, X. Gene transcripts in spermatozoa: Markers of male infertility. Clin. Chim. Acta 2012, 413, 1035–1038. [Google Scholar] [CrossRef] [PubMed]
- Coutton, C.; Escoffier, J.; Martinez, G.; Arnoult, C.; Ray, P.F. Teratozoospermia: Spotlight on the main genetic actors in the human. Hum. Reprod. Update 2015, 21, 455–485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.Y.; Lai, T.H.; Chen, M.F.; Lee, H.L.; Kuo, P.L.; Lin, Y.H. SEPT14 Mutations and Teratozoospermia: Genetic Effects on Sperm Head Morphology and DNA Integrity. J. Clin. Med. 2019, 8, 1297. [Google Scholar] [CrossRef] [PubMed]
- Omolaoye, T.S.; Hachim, M.Y.; du Plessis, S.S. Using publicly available transcriptomic data to identify mechanistic and diagnostic biomarkers in azoospermia and overall male infertility. Sci. Rep. 2022, 12, 2584. [Google Scholar] [CrossRef]
- Sánchez-Peña, M.L.; Isaza, C.E.; Pérez-Morales, J.; Rodríguez-Padilla, C.; Castro, J.M.; Cabrera-Ríos, M. Identification of potential biomarkers from microarray experiments using multiple criteria optimization. Cancer Med. 2013, 2, 253–265. [Google Scholar] [CrossRef]
- Juanes-Velasco, P.; Carabias-Sanchez, J.; Garcia-Valiente, R.; Fernandez-García, J.; Gongora, R.; Gonzalez-Gonzalez, M.; Fuentes, M. Microarrays as platform for multiplex assays in biomarker and drug discovery. In Rapid Test - Advances in Design, Format and Diagnostic Applications; IntechOpen: London, UK, 2018. [Google Scholar]
- Bottero, V.; Potashkin, J.A. Meta-analysis of gene expression changes in the blood of patients with mild cognitive impairment and alzheimer’s disease dementia. Int. J. Mol. Sci. 2019, 20, 5403. [Google Scholar] [CrossRef]
- Edgar, R.; Domrachev, M.; Lash, A.E. Gene expression omnibus: Ncbi gene expression and hybridization array data repository. Nucleic Acids Res. 2002, 30, 207–210. [Google Scholar] [CrossRef]
- Aponte, P.M. Spermatogonial stem cells: Current biotechnological advances in reproduction and regenerative medicine. World J. Stem Cells 2015, 7, 669–680. [Google Scholar] [CrossRef]
- Platts, A.E.; Dix, D.J.; Chemes, H.E.; Thompson, K.E.; Goodrich, R.; Rockett, J.C.; Rawe, V.Y.; Quintana, S.; Diamond, M.P.; Strader, L.F.; et al. Success and failure in human spermatogenesis as revealed by teratozoospermic rnas. Hum. Mol. Gen. 2007, 16, 763–773. [Google Scholar] [CrossRef]
- Hodžić, A.; Maver, A.; Plaseska-Karanfilska, D.; Ristanović, M.; Noveski, P.; Zorn, B.; Terzic, M.; Kunej, T.; Peterlin, B. De novo mutations in idiopathic male infertility-a pilot study. Andrology 2021, 9, 212–220. [Google Scholar] [CrossRef]
- Hadziselimovic, F.; Hadziselimovic, N.O.; Demougin, P.; Oakeley, E.J. Testicular gene expression in cryptorchid boys at risk of azoospermia. Sex. Dev. Genet. Mol. Biol. Evol. Endocrinol. Embryol. Pathol. Sex Determ. Differ. 2011, 5, 49–59. [Google Scholar] [CrossRef] [PubMed]
- Sharov, A.A.; Schlessinger, D.; Ko, M.S. Exatlas: An interactive online tool for meta-analysis of gene expression data. J. Bioinform. Comput. Biol. 2015, 13, 1550019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Cook, B.; Manning, W.; Alegria, M. Measuring disparities across the distribution of mental health care expenditures. J. Ment. Health Policy Econ. 2013, 16, 3–12. [Google Scholar] [PubMed]
- Zhou, G.; Soufan, O.; Ewald, J.; Hancock, R.E.W.; Basu, N.; Xia, J. Networkanalyst 3.0: A visual analytics platform for comprehensive gene expression profiling and meta-analysis. Nucleic Acids Res. 2019, 47, W234–W241. [Google Scholar] [CrossRef]
- Klemmt, P.A.B.; Starzinski-Powitz, A. Molecular and cellular pathogenesis of endometriosis. Curr. Women’s Health Rev. 2018, 14, 106–116. [Google Scholar] [CrossRef]
- Bell, A.; Fairbrother, M.; Jones, K. Fixed and random effects models: Making an informed choice. Qual. Quant. 2019, 53, 1051–1074. [Google Scholar] [CrossRef]
- Guha, P.; Roychoudhury, S.; Singha, S.; Kalita, J.C.; Kolesarova, A.; Jamal, Q.M.S.; Jha, N.K.; Kumar, D.; Ruokolainen, J.; Kesari, K.K. A comparative cross-platform meta-analysis to identify potential biomarker genes common to endometriosis and recurrent pregnancy loss. Appl. Sci. 2021, 11, 3349. [Google Scholar] [CrossRef]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. Limma powers differential expression analyses for rna-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Laganà, A.S.; Garzon, S.; Götte, M.; Viganò, P.; Franchi, M.; Ghezzi, F.; Martin, D.C. The pathogenesis of endometriosis: Molecular and cell biology insights. Int. J. Mol. Sci. 2019, 20, 5615. [Google Scholar] [CrossRef]
- Green, G.H.; Diggle, P.J. On the operational characteristics of the benjamini and hochberg false discovery rate procedure. Stat. Appl. Gen. Mol. Biol. 2007, 6, 27. [Google Scholar] [CrossRef]
- Mudunuri, U.; Che, A.; Yi, M.; Stephens, R.M. Biodbnet: The biological database network. Bioinformatics 2009, 25, 555–556. [Google Scholar] [CrossRef] [PubMed]
- Gu, Z.; Eils, R.; Schlesner, M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 2016, 32, 2847–2849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doncheva, N.T.; Morris, J.H.; Gorodkin, J.; Jensen, L.J. Cytoscape stringapp: Network analysis and visualization of proteomics data. J. Proteome Res. 2019, 18, 623–632. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Maere, S.; Heymans, K.; Kuiper, M. Bingo: A cytoscape plugin to assess overrepresentation of gene ontology categories in biological networks. Bioinformatics 2005, 21, 3448–3449. [Google Scholar] [CrossRef]
- Ignatieva, E.V.; Osadchuk, A.V.; Kleshchev, M.A.; Bogomolov, A.G.; Osadchuk, L.V. A catalog of human genes associated with pathozoospermia and functional characteristics of these genes. Front. Genet. 2021, 12, 662770. [Google Scholar] [CrossRef]
- Sengupta, P.; Cho, C.L. The pathophysiology of male infertility. In Male Infertility in Reproductive Medicine; CRC Press: Boca Raton, FL, USA, 2019; pp. 1–9. [Google Scholar]
- Krausz, C.; Riera-Escamilla, A. Genetics of male infertility. Nat. Rev. Urol. 2018, 15, 369–384. [Google Scholar] [CrossRef]
- World Health Organization. World Health Statistics; World Health Organization: Geneva, Switzerland, 2009. [Google Scholar]
- Chemes, E.H.; Rawe, Y.V. Sperm pathology: A step beyond descriptive morphology. Origin, characterization and fertility potential of abnormal sperm phenotypes in infertile men. Hum. Reprod. Update 2003, 9, 405–428. [Google Scholar] [CrossRef]
- Chemes, H.E.; Alvarez Sedo, C. Tales of the tail and sperm headaches: Changing concepts on the prognostic significance of sperm pathologies affecting the head, neck and tail. Asian J. Androl. 2012, 14, 14–23. [Google Scholar] [CrossRef]
- Malcher, A.; Rozwadowska, N.; Stokowy, T.; Kolanowski, T.; Jedrzejczak, P.; Zietkowiak, W.; Kurpisz, M. Potential biomarkers of nonobstructive azoospermia identified in microarray gene expression analysis. Fertil. Steril. 2013, 100, e1681–e1687. [Google Scholar] [CrossRef]
- Hu, T.; Luo, S.; Xi, Y.; Tu, X.; Yang, X.; Zhang, H.; Feng, J.; Wang, C.; Zhang, Y. Integrative bioinformatics approaches for identifying potential biomarkers and pathways involved in non-obstructive azoospermia. Transl. Androl. Urol. 2021, 10, 243–257. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Xie, N.; Xie, D.; Sun, L.; Li, S.; Li, P.; Li, Y.; Li, J.; Dong, Z.; Xie, X. A novel homozygous fbxo43 mutation associated with male infertility and teratozoospermia in a consanguineous chinese family. Fertil. Steril. 2019, 111, 909–917.e901. [Google Scholar] [CrossRef] [Green Version]
- Coutton, C.; Martinez, G.; Kherraf, Z.E.; Amiri-Yekta, A.; Boguenet, M.; Saut, A.; He, X.; Zhang, F.; Cristou-Kent, M.; Escoffier, J.; et al. Bi-allelic mutations in armc2 lead to severe astheno-teratozoospermia due to sperm flagellum malformations in humans and mice. Am. J. Hum. Gen. 2019, 104, 331–340. [Google Scholar] [CrossRef]
- Kuo, Y.C.; Lin, Y.H.; Chen, H.I.; Wang, Y.Y.; Chiou, Y.W.; Lin, H.H.; Pan, H.A.; Wu, C.M.; Su, S.M.; Hsu, C.C.; et al. Sept12 mutations cause male infertility with defective sperm annulus. Hum. Mutat. 2012, 33, 710–719. [Google Scholar] [CrossRef] [PubMed]
- Kastner, S.; Thiemann, I.J.; Dekomien, G.; Petrasch-Parwez, E.; Schreiber, S.; Akkad, D.A.; Gerding, W.M.; Hoffjan, S.; Günes, S.; Günes, S.; et al. Exome sequencing reveals agbl5 as novel candidate gene and additional variants for retinitis pigmentosa in five turkish families. Investig. Ophthal. Mol. Vis. Sci. 2015, 56, 8045–8053. [Google Scholar] [CrossRef] [PubMed]
- Kherraf, Z.E.; Cazin, C.; Lestrade, F.; Muronova, J.; Coutton, C.; Arnoult, C.; Thierry-Mieg, N.; Ray, P.F. From azoospermia tomacrozoospermia, a phenotypic continuum due to mutations in the ZMYND15 gene. Asian J. Androl. 2022, 24, 243–247. [Google Scholar]
- Wen, Y.; Wang, X.; Zheng, R.; Dai, S.; Li, J.; Yang, Y.; Shen, Y. Sequencing of the ZMYND15 gene in a cohort of infertile Chinese men reveals novel mutations in patients with teratozoospermia. J. Med. Genet. 2022, 108727. [Google Scholar] [CrossRef]
- Rousseaux-Prévost, R.; Lesur, P.; Collier, F.; Rigot, J.M.; Dalla Venezia, N.; Pol, P.S.; Delaunay, J.; Gauthier, A.; Rousseaux, J. Abnormal expression of protein 4.1 in spermatozoa of infertile men with teratospermia. Lancet 1994, 343, 764–765. [Google Scholar] [CrossRef]
- Chen, J.; Gu, Y.; Zhang, Z.; Zheng, W.; Yang, L.; Huang, W.; Lin, S.; Li, Y.; Guo, H.; Luo, M.; et al. Deficiency of spata46, a novel nuclear membrane protein, causes subfertility in male mice. Biol. Reprod. 2016, 95, 58. [Google Scholar] [CrossRef]
- Zhou, J.H.; Zhou, Q.Z.; Lyu, X.M.; Zhu, T.; Chen, Z.J.; Chen, M.K.; Xia, H.; Wang, C.Y.; Qi, T.; Li, X.; et al. The expression of cysteine-rich secretory protein 2 (crisp2) and its specific regulator mir-27b in the spermatozoa of patients with asthenozoospermia. Biol. Reprod. 2015, 92, 28. [Google Scholar] [CrossRef]
- Yuan, S.; Stratton, C.J.; Bao, J.; Zheng, H.; Bhetwal, B.P.; Yanagimachi, R.; Yan, W. Spata6 is required for normal assembly of the sperm connecting piece and tight head-tail conjunction. Proc. Natl. Acad. Sci. USA 2015, 112, E430–E439. [Google Scholar] [CrossRef] [PubMed]
- Bracke, A.; Peeters, K.; Punjabi, U.; Hoogewijs, D.; Dewilde, S. A search for molecular mechanisms underlying male idiopathic infertility. Reprod. Biomed. Online 2018, 36, 327–339. [Google Scholar] [CrossRef] [PubMed]
- Fox, M.S.; Ares, V.X.; Turek, P.J.; Haqq, C.; Reijo Pera, R.A. Feasibility of global gene expression analysis in testicular biopsies from infertile men. Mol. Reprod. Dev. 2003, 66, 403–421. [Google Scholar] [CrossRef] [PubMed]
- Ellis, P.J.; Furlong, R.A.; Conner, S.J.; Kirkman-Brown, J.; Afnan, M.; Barratt, C.; Griffin, D.K.; Affara, N.A. Coordinated transcriptional regulation patterns associated with infertility phenotypes in men. J. Med. Genet. 2007, 44, 498–508. [Google Scholar] [CrossRef]
- Spiess, A.N.; Feig, C.; Schulze, W.; Chalmel, F.; Cappallo-Obermann, H.; Primig, M.; Kirchhoff, C. Cross-platform gene expression signature of human spermatogenic failure reveals inflammatory-like response. Hum. Reprod. 2007, 22, 2936–2946. [Google Scholar] [CrossRef]
- Cooke, H.; Hargreave, T.; Elliott, D. Understanding the genes involved in spermatogenesis: A progress report. Fertil. Steril. 1998, 69, 989–995. [Google Scholar] [CrossRef]
- Grootegoed, J.A.; Siep, M.; Baarends, W.M. Molecular and cellular mechanisms in spermatogenesis. Best Pract. Res. Clin. Endocrinol. 2000, 14, 331–343. [Google Scholar] [CrossRef]
- Johnson, L. Efficiency of spermatogenesis. Micros. Res. Tech. 1995, 3, 385–422. [Google Scholar] [CrossRef]
- Paquis-Flucklinger, V.; Santucci-Darmanin, S.; Paul, R.; Saunieres, A.; Turc-Carel, C.; Desnuelle, C. Cloning and expression analysis of a meiosis-specific MutS homolog: The human MSH4 gene. Genomics 1997, 44, 188–194. [Google Scholar] [CrossRef]
- Lundgren, K.; Walworth, N.; Booher, R.; Dembski, M.; Kirschner, M.; Beach, D. mik1 and wee1 cooperate in the inhibitory tyrosine phosphorylation of cdc2. Cell 1991, 64, 1111–1122. [Google Scholar] [CrossRef]
- Subramaniam, K.; Seydoux, G. nos-1 and nos-2, two genes related to Drosophila nanos, regulate primordial germ cell development and survival in Caenorhabditis elegans. Development 1999, 126, 4861–4871. [Google Scholar] [CrossRef] [PubMed]
- Subramaniam, K.; Seydoux, G. Dedifferentiation of primary spermatocytes into germ cell tumors in C. elegans lacking the pumilio-like protein PUF-8. Curr. Biol. 2003, 13, 134–139. [Google Scholar] [PubMed]
- Dada, R.; Ahmed, M.E.; Talwar, R.; Kucheria, K. Clinical and Genetic study in a XX (SRY negative) male. Int. J. Med. 2002. [Google Scholar]
- Shamsi, M.B.; Kumar, K.; Dada, R. Genetic and epigenetic factors: Role in male infertility. Indian J. Urol. 2011, 27, 110–120. [Google Scholar] [PubMed]
- Boué, A.; Gallano, P. A collaborative study of the segregation of inherited chromosome structural rearrangements in 1356 prenatal diagnoses. Prenat. Diagn. 1984, 4, 45–67. [Google Scholar] [CrossRef]
- Rodríguez de la Vega Otazo, M.; Lorenzo, J.; Tort, O.; Avilés, F.X.; Bautista, J.M. Functional segregation and emerging role of cilia-related cytosolic carboxypeptidases (CCPs). FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2013, 27, 424–431. [Google Scholar]
- Rogowski, K.; van Dijk, J.; Magiera, M.M.; Bosc, C.; Deloulme, J.C.; Bosson, A.; Peris, L.; Gold, N.D.; Lacroix, B.; Bosch Grau, M.; et al. A family of protein-deglutamylating enzymes associated with neurodegeneration. Cell 2010, 143, 564–578. [Google Scholar] [CrossRef]
- Wu, H.Y.; Rong, Y.; Correia, K.; Min, J.; Morgan, J.I. Comparison of the enzymatic and functional properties of three cytosolic carboxypeptidase family members. J. Biol. Chem. 2015, 290, 1222–1232. [Google Scholar] [CrossRef]
- Carrell, D.T. Methods of Identifying Male Fertility Status and Embryo Quality. U.S. Patent Application No. 15/750,715, 19 April 2019. [Google Scholar]
- Lea, I.A.; Richardson, R.T.; Widgren, E.E.; O’Rand, M.G. Cloning and sequencing of cdnas encoding the human sperm protein, sp17. Biochim. Biophys. Acta 1996, 1307, 263–266. [Google Scholar] [CrossRef]
- Gjerstorff, M.F.; Andersen, M.H.; Ditzel, H.J. Oncogenic cancer/testis antigens: Prime candidates for immunotherapy. Oncotarget 2015, 6, 15772–15787. [Google Scholar] [CrossRef]
- Wen, Y.; Richardson, R.T.; Widgren, E.E.; O’Rand, M.G. Characterization of sp17: A ubiquitous three domain protein that binds heparin. Biochem 2001, 357, 25–31. [Google Scholar] [CrossRef]
- Zhang, Q.; Gao, M.; Zhang, Y.; Song, Y.; Cheng, H.; Zhou, R. The germline-enriched Ppp1r36 promotes autophagy. Sci. Rep. 2016, 6, 24609. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Kuang, J.; Zhong, L.; Kuo, W.L.; Gray, J.W.; Sahin, A.; Brinkley, B.R.; Sen, S. Tumour amplified kinase STK15/BTAK induces centrosome amplification, aneuploidy and transformation. Nat. Genet. 1998, 20, 189–193. [Google Scholar] [CrossRef] [PubMed]
- Crane, R.; Gadea, B.; Littlepage, L.; Wu, H.; Ruderman, J.V. Aurora A, meiosis and mitosis. Biol. Cell 2004, 96, 215–229. [Google Scholar] [CrossRef]
- Roig, I.; Dowdle, J.A.; Toth, A.; de Rooij, D.G.; Jasin, M.; Keeney, S. Mouse TRIP13/PCH2 is required for recombination and normal higher-order chromosome structure during meiosis. PLoS Genet. 2010, 6, e1001062. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.T.; Poon, R. TRIP13 Regulates Both the Activation and Inactivation of the Spindle-Assembly Checkpoint. Cell Rep. 2016, 14, 1086–1099. [Google Scholar] [CrossRef]
- Vader, G. Pch2(TRIP13): Controlling cell division through regulation of HORMA domains. Chromosoma 2015, 124, 333–339. [Google Scholar] [CrossRef]
- Archambault, V.; Pinson, X. Free centrosomes: Where do they all come from? Fly 2010, 4, 172–177. [Google Scholar] [CrossRef]
- Barr, F.A.; Silljé, H.H.; Nigg, E.A. Polo-like kinases and the orchestration of cell division. Nat. Rev. Mol. Cell Biol. 2004, 5, 429–440. [Google Scholar] [CrossRef]
- Adlakha, J.; Karamichali, I.; Sangwallek, J.; Deiss, S.; Bär, K.; Coles, M.; Hartmann, M.D.; Lupas, A.N.; Hernandez Alvarez, B. Characterization of mcu-binding proteins mcur1 and ccdc90b—representatives of a protein family conserved in prokaryotes and eukaryotic organelles. Structure 2019, 27, 464–475.e466. [Google Scholar] [CrossRef] [Green Version]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sl. No. | GEO Accession | Subject | Sample | Analytical Platform | Patient Type | Ref. | ||
|---|---|---|---|---|---|---|---|---|
| Patient | Control | Total | ||||||
| 1 | GSE6872 | 8 | 13 | 21 | Spermatozoa | GPL570 ([HG-U133_Plus_2] Affymetrix Human Genome U133 Plus 2.0 Array) | Teratozoospermia | [30] |
| 2 | GSE6967 | 8 | 5 | 13 | Spermatozoa | GPL2507 (Sentrix Human-6 Expression BeadChip) | Teratozoospermia | [30] |
| 3 | GSE145467 | 10 | 10 | 20 | Testis tissue | GPL4133 (Agilent-014850 Whole Human Genome Microarray 4x44K G4112F (Feature Number Version)) | Azoospermia | [31] |
| 4 | GSE25518 | 19 | 4 | 23 | Testis tissue | GPL570 ([HG-U133_Plus_2] Affymetrix Human Genome U133 Plus 2.0 Array) | Azoospermia | [32] |
| Gene Symbol | Entrez ID | Log Ratio Combined | Fold Change | FDR |
|---|---|---|---|---|
| SPA17 | Sperm autoantigenic protein 17 | −0.9764 | 9.471 | 0.0366 |
| TKTL1 | Transketolase-like 1 | −0.9288 | 8.488 | 1.01 × 10−7 |
| DDX43 | DEAD (Asp-Glu-Ala-Asp) box polypeptide 43 | −0.8869 | 7.708 | 0.014 |
| PRKAA1 | Protein kinase, AMP-activated, α 1 catalytic subunit | −0.8464 | 7.021 | 0.000101 |
| SPATA22 | Spermatogenesis associated 22 | −0.8169 | 6.561 | 0.028 |
| HNRNPM | Heterogeneous nuclear ribonucleoprotein M | −0.7491 | 5.612 | 4.31 × 10−6 |
| TPTE | Transmembrane phosphatase with tensin homology | −0.7235 | 5.291 | 2.65 × 10−7 |
| EIF4A2 | Eukaryotic translation initiation factor 4A2 | −0.7106 | 5.136 | 0.0251 |
| UBE2D3 | Ubiquitin-conjugating enzyme E2D 3 | −0.6681 | 4.656 | 4.65 × 10−7 |
| ADAMTS5 | ADAM metallopeptidase with thrombospondin type 1 motif 5 | 0.6675 | 4.65 | 0.0446 |
| OSBPL10 | Oxysterol binding protein-like 10 | −0.6591 | 4.561 | 0.001776 |
| EFHC1 | EF-hand domain (C-terminal) containing 1 | −0.6357 | 4.322 | 0.009339 |
| DLGAP5 | Discs, large (Drosophila) homolog-associated protein 5 | −0.6311 | 4.276 | 0.003758 |
| PPP1R36 | Protein phosphatase 1 regulatory subunit 36 | −0.6296 | 4.262 | 0.004044 |
| TAF5 | TATA-box binding protein associated factor 5 | −0.613 | 4.102 | 0.004949 |
| GTF2A2 | General transcription factor IIA 2 | −0.6102 | 4.076 | 1.13 × 10−6 |
| PARM1 | Prostate androgen-regulated mucin-like protein 1 | 0.602 | 3.999 | 0.000384 |
| REXO5 | RNA exonuclease 5 | −0.5984 | 3.967 | 0.008627 |
| CDCA2 | Cell division cycle associated 2 | −0.5947 | 3.933 | 0.0338 |
| CLDN5 | Claudin 5 | 0.5839 | 3.836 | 0.0363 |
| DGAT2 | Diacylglycerol O-acyltransferase 2 | −0.5793 | 3.796 | 0.000383 |
| PLK4 | Polo-like kinase 4 | −0.5708 | 3.722 | 0.0456 |
| RALGPS2 | Ral GEF with PH domain and SH3 binding motif 2 | −0.5698 | 3.714 | 7.50 × 10−6 |
| KIF2C | Kinesin family member 2C | −0.56 | 3.631 | 3.96 × 10−6 |
| RACGAP1 | Rac GTPase activating protein 1 | −0.5592 | 3.624 | 0.0247 |
| Name | Average Shortest Path Length | Betweenness Centrality | Closeness Centrality | Clustering Coefficient | Degree |
|---|---|---|---|---|---|
| AURKA | 2.634615 | 0.268733 | 0.379562 | 0.538462 | 14 |
| TRIP13 | 2.557692 | 0.383003 | 0.390977 | 0.615385 | 13 |
| PLK4 | 2.865385 | 0.053786 | 0.348993 | 0.621212 | 12 |
| DLGAP5 | 2.846154 | 0.078277 | 0.351351 | 0.727273 | 12 |
| RACGAP1 | 2.884615 | 0.002863 | 0.346667 | 0.872727 | 11 |
| KIF2C | 2.884615 | 0.002863 | 0.346667 | 0.872727 | 11 |
| DTL | 2.807692 | 0.11128 | 0.356164 | 0.745455 | 11 |
| CDCA2 | 2.903846 | 0.021237 | 0.344371 | 0.781818 | 11 |
| KPNA2 | 2.942308 | 0.019194 | 0.339869 | 0.777778 | 10 |
| TACC3 | 3.038462 | 0.038629 | 0.329114 | 0.75 | 9 |
| MND1 | 2.961538 | 1.68 × 10−4 | 0.337662 | 0.972222 | 9 |
| ATAD2 | 2.980769 | 0.038462 | 0.335484 | 0.75 | 8 |
| CAV1 | 3.480769 | 0.248291 | 0.287293 | 0.066667 | 6 |
| DDX3X | 3.211538 | 0.180979 | 0.311377 | 0.2 | 6 |
| EIF4A2 | 3.076923 | 0.194051 | 0.325 | 0.2 | 5 |
| CASP1 | 4.019231 | 0.147059 | 0.248804 | 0.166667 | 4 |
| SYCP3 | 3.346154 | 0.180241 | 0.298851 | 0.166667 | 4 |
| TRIP12 | 3.5 | 0.090196 | 0.285714 | 0.166667 | 4 |
| TEX15 | 4.230769 | 0.076169 | 0.236364 | 0.166667 | 4 |
| SDPR | 4.211538 | 0.03449 | 0.237443 | 0.333333 | 3 |
| GO ID | Gene Names | Description | Average Shortest Path Length | Betweenness Centrality | Closeness Centrality | Neighborhood Connectivity | Node Size | No. of Genes | Adjusted p-Value |
|---|---|---|---|---|---|---|---|---|---|
| 7049 | STEAP3, CDCA2, CUL2, SIAH1, TUBE1, MND1, TEX15, AURKA, RACGAP1, SPIN1, TACC3, KIF2C, TRIP13, SYCP3, KPNA2, and DLGAP5 | Cell cycle | 2.25 | 0.176768 | 0.444444 | 3 | 8 | 16 | 1.57 × 10−2 |
| 22403 | CDCA2, CUL2, TACC3, KIF2C, MND1, TEX15, TRIP13, SYCP3, KPNA2, DLGAP5, and AURKA | Cell cycle phase | 2.333333 | 0.320707 | 0.428571 | 2.666667 | 6.63325 | 11 | 1.57 × 10−2 |
| 22402 | CDCA2, CUL2, TUBE1, MND1, TEX15, AURKA, RACGAP1, TACC3, KIF2C, TRIP13, SYCP3, KPNA2, and DLGAP5 | Cell cycle process | 2.083333 | 0.34596 | 0.48 | 3.333333 | 7.211103 | 13 | 1.57 × 10−2 |
| 279 | CDCA2, TACC3, KIF2C, MND1, TEX15, TRIP13, SYCP3, KPNA2, DLGAP5, and AURKA | M phase | 2.916667 | 0.017677 | 0.342857 | 3 | 6.324555 | 10 | 1.57 × 10−2 |
| 15630 | PLK4, RANBP9, TUBE1, AURKA, RACGAP1, CSPP1, DLC1, VPS41, SPIN1, TACC3, KIF2C, DTL, and DLGAP5 | Microtubule cytoskeleton | 2.85 | 0.014912 | 0.350877 | 3.666667 | 7.211103 | 13 | 1.57 × 10−2 |
| 9987 | EIF4A2, STEAP3, SPON1, USPL1, HHIP, UBE2D3, STON1, AREG, BAIAP2L1, CSRP2, TOP1MT, SPIN1, CASP1, QSOX1, KPNA2, SPA17, STK32B, DLGAP5, VAV3, TOR3A, TKTL1, DGAT2, PTGIS, METTL3, VPS13A, ATP1B2, TEX15, PASK, PIAS2, CLDN5CCDC80, HAND2, NUP50, ADPRH, RHOJ, FAR1, KIF2C, RPP25, DTL, GTF2A2, PRKAA1, CDCA2, CUL2, GMPS, TPTE, CYP17A1, AURKA, AP3M2, WNT6, PPP1R2, RACGAP1, STK36, EPB41L3, PLEK2, PLK4, XRCC6, CAV1, SIAH1, FMO1, RANBP9, TUBE1, MND1, SNAP91, HNRNPM, DLC1, MGAT4A, VPS41, TACC3, TRIP12, TAF5, TRIP13, SYCP3, and EIF4G3 | Cellular process | 2.083333 | 0.5 | 0.48 | 2.75 | 17.08801 | 73 | 3.59 × 10−2 |
| 5813 | PLK4, TACC3, TUBE1, DTL, DLGAP5, and AURKA | Centrosome | 2.6 | 0.078421 | 0.384615 | 3.666667 | 4.898979 | 6 | 4.92 × 10−2 |
| 31616 | DLGAP5 and AURKA | Spindle pole centrosome | 3.25 | 0.005263 | 0.307692 | 3 | 2.828427 | 2 | 4.92 × 10−2 |
| 6996 | VAV3, XRCC6, CDCA2, CAV1, RANBP9, TUBE1, TEX15, SNAP91, AURKA, RACGAP1, DLC1, EPB41L3, RHOJ, TACC3, PLEK2, KIF2C, TAF5, SYCP3, and DLGAP5 | Organelle organization | 2.666667 | 0.234848 | 0.375 | 2.333333 | 8.717798 | 19 | 4.92 × 10−2 |
| 7010 | RACGAP1, DLC1, EPB41L3, RHOJ, TACC3, RANBP9, PLEK2, KIF2C, TUBE1, and AURKA | Cytoskeleton organization | 3.583333 | 0 | 0.27907 | 3 | 6.324555 | 10 | 4.92 × 10−2 |
| 7140 | TEX15, TRIP13, and SYCP3 | Male meiosis | 3.666667 | 0 | 0.272727 | 3 | 3.464102 | 3 | 4.92 × 10−2 |
| 8022 | XRCC6, RACGAP1, CAV1, DLC1, SIAH1, and EFHC1 | Protein C-terminus binding | 2 | 0 | 0.5 | 2 | 4.898979486 | 3 | 4.92 × 10−2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Das, S.; Guha, P.; Nath, M.; Das, S.; Sen, S.; Sahu, J.; Kopanska, M.; Dutta, S.; Jamal, Q.M.S.; Kesari, K.K.; et al. A Comparative Cross-Platform Analysis to Identify Potential Biomarker Genes for Evaluation of Teratozoospermia and Azoospermia. Genes 2022, 13, 1721. https://doi.org/10.3390/genes13101721
Das S, Guha P, Nath M, Das S, Sen S, Sahu J, Kopanska M, Dutta S, Jamal QMS, Kesari KK, et al. A Comparative Cross-Platform Analysis to Identify Potential Biomarker Genes for Evaluation of Teratozoospermia and Azoospermia. Genes. 2022; 13(10):1721. https://doi.org/10.3390/genes13101721
Chicago/Turabian StyleDas, Suchismita, Pokhraj Guha, Monika Nath, Sandipan Das, Surojit Sen, Jagajjit Sahu, Marta Kopanska, Sulagna Dutta, Qazi Mohammad Sajid Jamal, Kavindra Kumar Kesari, and et al. 2022. "A Comparative Cross-Platform Analysis to Identify Potential Biomarker Genes for Evaluation of Teratozoospermia and Azoospermia" Genes 13, no. 10: 1721. https://doi.org/10.3390/genes13101721