
Individual Identification with Short Tandem Repeat Analysis and Collection of Secondary Information Using Microbiome Analysis

 , ,
, ,  , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
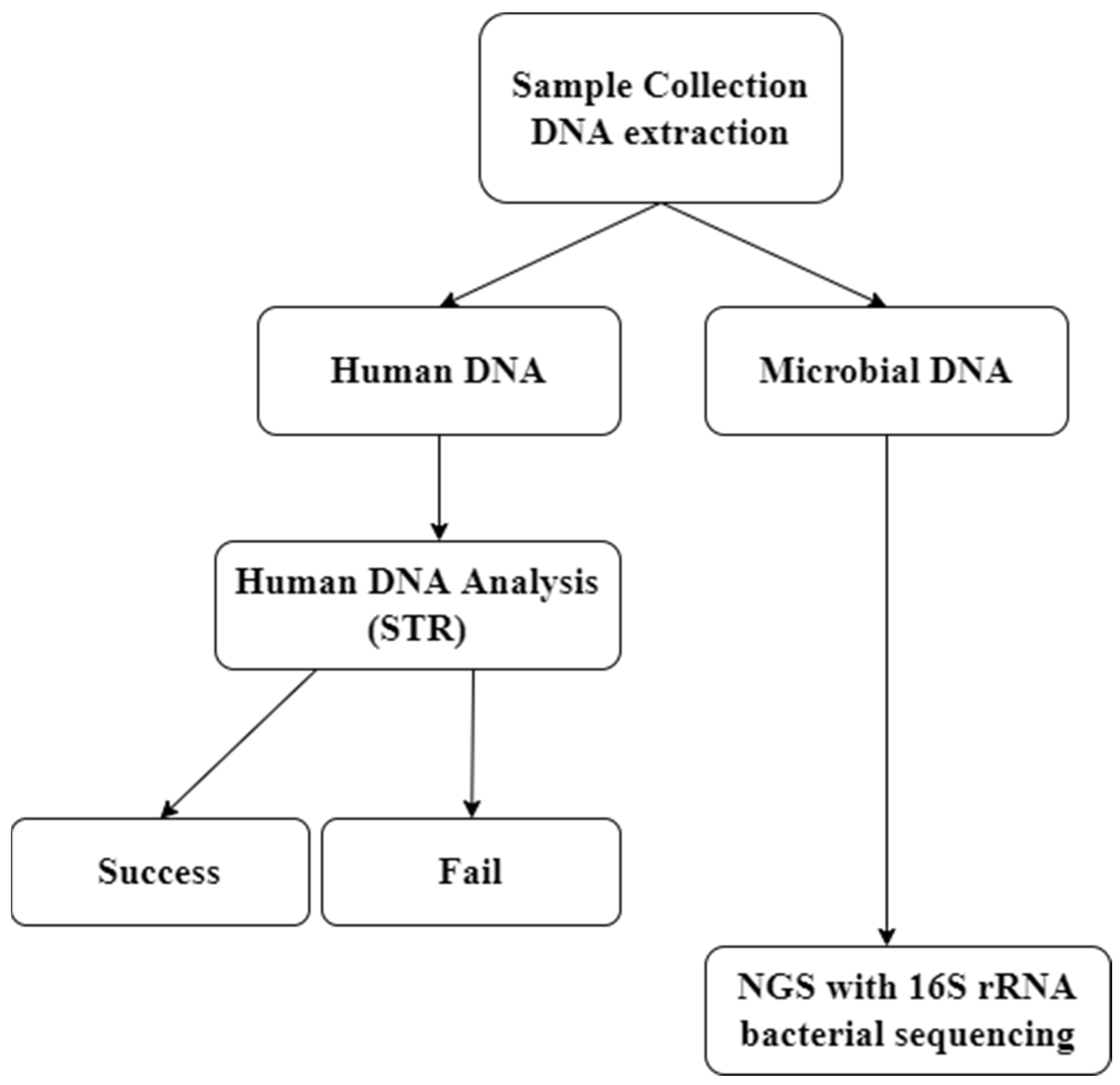
2.1. Sample Collection and DNA Extraction
2.2. STR Analysis
2.3. Library Preparation of 16S rRNA Amplicons
2.4. Analysis of 16S rRNA Amplicon Sequences
2.5. Statistical Analysis
3. Results
3.1. STR Analysis
3.2. Microbiome Analysis
3.2.1. Overview of Taxonomic Diversity
3.2.2. α and β Diversities
3.2.3. Creation of a List of Strains and Matching Assessment
3.3. Personal Feature Tracking and Unique Bacterial Features
3.4. Bacterial Analysis of Failed STR an Analysis Samples
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Reid, C.A.; Howes, L.M. Communicating forensic scientific expertise: An analysis of expert reports and corresponding testimony in Tasmanian courts. Sci. Justice 2020, 60, 108–119. [Google Scholar] [CrossRef] [PubMed]
- Dumache, R.; Ciocan, V.; Muresan, C.; Enache, A. Molecular DNA Analysis in Forensic Identification. Clin. Lab. 2016, 62, 245–248. [Google Scholar] [CrossRef] [PubMed]
- Silva, N.M.; Pereira, L.; Poloni, E.S.; Currat, M. Human neutral genetic variation and forensic STR data. PLoS ONE 2012, 7, e49666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dziak, R.; Peneder, A.; Buetter, A.; Hageman, C. Trace DNA Sampling Success from Evidence Items Commonly Encountered in Forensic Casework. J. Forensic Sci. 2018, 63, 835–841. [Google Scholar] [CrossRef] [PubMed]
- Raymond, J.J.; van Oorschot, R.A.; Walsh, S.J.; Roux, C. Trace DNA analysis: Do you know what your neighbour is doing? A multi-jurisdictional survey. Forensic Sci. Int. Genet. 2008, 2, 19–28. [Google Scholar] [CrossRef]
- Harbison, S.; Fallow, M.; Bushell, D. An analysis of the success rate of 908 trace DNA samples submitted to the Crime Sample Database Unit in New Zealand. Aust. J. Forensic Sci. 2008, 40, 49–53. [Google Scholar] [CrossRef]
- Alaeddini, R.; Walsh, S.J.; Abbas, A. Forensic implications of genetic analyses from degraded DNA—A review. Forensic Sci. Int. Genet. 2010, 4, 148–157. [Google Scholar] [CrossRef]
- Kloosterman, A.; Kersbergen, P. Efficacy and limits of genotyping low copy number DNA samples by multiplex PCR of STR loci. Int. Congr. Ser. 2003, 1239, 795–798. [Google Scholar] [CrossRef]
- Meadow, J.F.; Altrichter, A.E.; Bateman, A.C.; Stenson, J.; Brown, G.Z.; Green, J.L.; Bohannan, B.J. Humans differ in their personal microbial cloud. PeerJ 2015, 3, e1258. [Google Scholar] [CrossRef] [Green Version]
- Eetemadi, A.; Rai, N.; Pereira, B.M.P.; Kim, M.; Schmitz, H.; Tagkopoulos, I. The Computational Diet: A Review of Computational Methods Across Diet, Microbiome, and Health. Front. Microbiol. 2020, 11, 393. [Google Scholar] [CrossRef] [Green Version]
- Pasolli, E.; Asnicar, F.; Manara, S.; Zolfo, M.; Karcher, N.; Armanini, F.; Beghini, F.; Manghi, P.; Tett, A.; Ghensi, P.; et al. Extensive Unexplored Human Microbiome Diversity Revealed by over 150,000 Genomes from Metagenomes Spanning Age, Geography, and Lifestyle. Cell 2019, 176, 649–662.e20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De la Cuesta-Zuluaga, J.; Kelley, S.T.; Chen, Y.; Escobar, J.S.; Mueller, N.T.; Ley, R.E.; McDonald, D.; Huang, S.; Swafford, A.D.; Knight, R.; et al. Age- and Sex-Dependent Patterns of Gut Microbial Diversity in Human Adults. mSystems 2019, 4, e00261-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huttenhower, C.; Gevers, D.; Knight, R.; Abubucker, S.; Badger, J.; Chinwalla, A.; Giglio, M.G. Structure, function and diversity of the healthy human microbiome. Nature 2012, 486, 207. [Google Scholar]
- Metcalf, J.L. Estimating the postmortem interval using microbes: Knowledge gaps and a path to technology adoption. Forensic Sci. Int. Genet. 2019, 38, 211–218. [Google Scholar] [CrossRef] [Green Version]
- Schmedes, S.E.; Woerner, A.E.; Novroski, N.M.M.; Wendt, F.R.; King, J.L.; Stephens, K.M.; Budowle, B. Targeted sequencing of clade-specific markers from skin microbiomes for forensic human identification. Forensic Sci. Int. Genet. 2018, 32, 50–61. [Google Scholar] [CrossRef]
- Fierer, N.; Lauber, C.L.; Zhou, N.; McDonald, D.; Costello, E.K.; Knight, R. Forensic identification using skin bacterial communities. Proc. Natl. Acad. Sci. USA 2010, 107, 6477–6481. [Google Scholar] [CrossRef] [Green Version]
- Goga, H. Comparison of bacterial DNA profiles of footwear insoles and soles of feet for the forensic discrimination of footwear owners. Int. J. Legal Med. 2012, 126, 815–823. [Google Scholar] [CrossRef]
- Yang, J.; Tsukimi, T.; Yoshikawa, M.; Suzuki, K.; Takeda, T.; Tomita, M.; Fukuda, S. Cutibacterium acnes (Propionibacterium acnes) 16S rRNA genotyping of microbial samples from possessions contributes to owner identification. mSystems 2019, 4, e00594-19. [Google Scholar] [CrossRef] [Green Version]
- Concheri, G.; Bertoldi, D.; Polone, E.; Otto, S.; Larcher, R.; Squartini, A. Chemical elemental distribution and soil DNA fingerprints provide the critical evidence in murder case investigation. PLoS ONE 2011, 6, e20222. [Google Scholar] [CrossRef]
- López, C.D.; González, D.M.; Haas, C.; Vidaki, A.; Kayser, M. Microbiome-based body site of origin classification of forensically relevant blood traces. Forensic Sci. Int. Genet. 2020, 47, 102280. [Google Scholar] [CrossRef] [PubMed]
- Aroutcheva, A.A.; Simoes, J.A.; Behbakht, K.; Faro, S. Gardnerella vaginalis isolated from patients with bacterial vaginosis and from patients with healthy vaginal ecosystems. Clin. Infect. Dis. 2001, 33, 1022–1027. [Google Scholar] [CrossRef] [Green Version]
- Alcaraz, L.D.; Belda-Ferre, P.; Cabrera-Rubio, R.; Romero, H.; Simón-Soro, A.; Pignatelli, M.; Mira, A. Identifying a healthy oral microbiome through metagenomics. Clin. Microbiol. Infect. 2012, 18 (Suppl. 4), 54–57. [Google Scholar] [CrossRef] [Green Version]
- Bisgaard, M.; Nørskov-Lauritsen, N.; De Wit, S.; Hess, C.; Christensen, H. Multilocus sequence phylogenetic analysis of Avibacterium. Microbiology 2012, 158, 993–1004. [Google Scholar] [CrossRef] [Green Version]
- Ho, W. What’s the Y-STR Gene? Determined for a 12-Year Sentence of Not Guilty. NEWSIS. 2020. Available online: https://news.v.daum.net/v/20200529142012559 (accessed on 29 May 2020).
- Lee, S.H.; You, H.S.; Kang, H.G.; Kang, S.S.; Hyun, S.H. Association between Altered Blood Parameters and Gut Microbiota after Synbiotic Intake in Healthy, Elderly Korean Women. Nutrients 2020, 12, 3112. [Google Scholar] [CrossRef] [PubMed]
- Dhariwal, A.; Chong, J.; Habib, S.; King, I.L.; Agellon, L.B.; Xia, J. MicrobiomeAnalyst: A web-based tool for comprehensive statistical, visual and meta-analysis of microbiome data. Nucleic Acids Res. 2017, 45, W180–W188. [Google Scholar] [CrossRef] [PubMed]
- Romsos, E.L.; French, J.L.; Smith, M.; Figarelli, V.; Harran, F.; Vandegrift, G.; Moreno, L.I.; Callaghan, T.F.; Brocato, J.; Vaidyanathan, J. Results of the 2018 rapid DNA maturity assessment. J. Forensic Sci. 2020, 65, 953–959. [Google Scholar] [CrossRef]
- Abelson, B.; Sun, D.; Que, L.; Nebel, R.A.; Baker, D.; Popiel, P.; Amundsen, C.L.; Chai, T.; Close, C.; DiSanto, M. Sex differences in lower urinary tract biology and physiology. Biol. Sex Differ. 2018, 9, 45. [Google Scholar] [CrossRef] [Green Version]
- Willis, J.R.; Gabaldón, T. The human oral microbiome in health and disease: From sequences to ecosystems. Microorganisms 2020, 8, 308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edmonds-Wilson, S.L.; Nurinova, N.I.; Zapka, C.A.; Fierer, N.; Wilson, M. Review of human hand microbiome research. J. Dermatol. Sci. 2015, 80, 3–12. [Google Scholar] [CrossRef] [Green Version]
- Ohta, J.; Sakurada, K. Oral gram-positive bacterial DNA-based identification of saliva from highly degraded samples. Forensic Sci. Int. Genet. 2019, 42, 103–112. [Google Scholar] [CrossRef]
- Petrova, M.I.; Lievens, E.; Malik, S.; Imholz, N.; Lebeer, S. Lactobacillus species as biomarkers and agents that can promote various aspects of vaginal health. Front. Physiol. 2015, 6, 81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Xia, J.; Jiang, L.; Tan, Y.; An, Y.; Zhu, X.; Ruan, J.; Chen, Z.; Zhen, H.; Ma, Y. Characterization of the human skin resistome and identification of two microbiota cutotypes. Microbiome 2021, 9, 47. [Google Scholar] [CrossRef]
- Killer, J.; Kopečný, J.; Mrázek, J.; Havlík, J.; Koppová, I.; Benada, O.; Rada, V.; Kofroňová, O. Bombiscardovia coagulans gen. nov., sp. nov., a new member of the family Bifidobacteriaceae isolated from the digestive tract of bumblebees. Syst. Appl. Microbiol. 2010, 33, 359–366. [Google Scholar] [CrossRef] [PubMed]
- Weon, H.-Y.; Kim, B.-Y.; Hong, S.-B.; Joa, J.-H.; Nam, S.-S.; Lee, K.H.; Kwon, S.-W. Skermanella aerolata sp. nov., isolated from air, and emended description of the genus Skermanella. Int. J. Syst. Evol. Microbiol. 2007, 57, 1539–1542. [Google Scholar] [CrossRef]
- Mitra, A.; MacIntyre, D.A.; Mahajan, V.; Lee, Y.S.; Smith, A.; Marchesi, J.R.; Lyons, D.; Bennett, P.R.; Kyrgiou, M. Comparison of vaginal microbiota sampling techniques: Cytobrush versus swab. Sci. Rep. 2017, 7, 9802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tozzo, P.; D’Angiolella, G.; Brun, P.; Castagliuolo, I.; Gino, S.; Caenazzo, L. Skin Microbiome analysis for forensic human identification: What do we know so far? Microorganisms 2020, 8, 873. [Google Scholar] [CrossRef]
- Thornbury, D.; Goray, M.; van Oorschot, R.A. Indirect DNA transfer without contact from dried biological materials on various surfaces. Forensic Sci. Int. Genet. 2021, 51, 102457. [Google Scholar] [CrossRef]
- Bondurant, S.W.; Duley, C.M.; Harbell, J.W. Demonstrating the persistent antibacterial efficacy of a hand sanitizer containing benzalkonium chloride on human skin at 1, 2, and 4 hours after application. Am. J. Infect. Control. 2019, 47, 928–932. [Google Scholar] [CrossRef] [Green Version]
- Tuzil, J.; Filkova, B.; Malina, J.; Kerestes, J.; Doležal, T. Smoking in women with chronic vaginal discomfort is not associated with decreased abundance of Lactobacillus spp. but promotes Mobiluncus and Gardnerella spp. overgrowth: Secondary analysis of trial data including microbiome analysis. Ceska Gynekol. 2021, 86, 22–29. [Google Scholar] [CrossRef]
- Dzidic, M.; Abrahamsson, T.R.; Artacho, A.; Collado, M.C.; Mira, A.; Jenmalm, M. Oral microbiota maturation during the first 7 years of life in relation to allergy development. Allergy 2018, 73, 2000–2011. [Google Scholar] [CrossRef] [Green Version]
- Du, Y.; Yu, X.; Wang, G. Massilia tieshanensis sp. nov., isolated from mining soil. Int. J. Syst. Evol. Microbiol. 2012, 62, 2356–2362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barka, E.A.; Vatsa, P.; Sanchez, L.; Gaveau-Vaillant, N.; Jacquard, C.; Klenk, H.-P.; Clément, C.; Ouhdouch, Y.; van Wezel, G.P. Taxonomy, physiology, and natural products of Actinobacteria. Microbiol. Mol. Biol. Rev. 2016, 80, 1–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hedman, J.; Jansson, L.; Akel, Y.; Wallmark, N.; Liljestrand, R.G.; Forsberg, C.; Ansell, R. The double-swab technique versus single swabs for human DNA recovery from various surfaces. Forensic Sci. Int. Genet. 2020, 46, 102253. [Google Scholar] [CrossRef] [PubMed]
- You, H.S.; Lee, S.H.; Ok, Y.J.; Kang, H.-G.; Sung, H.J.; Lee, J.Y.; Kang, S.S.; Hyun, S.H. Influence of swabbing solution and swab type on DNA recovery from rigid environmental surfaces. J. Microbiol. Methods 2019, 161, 12–17. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Sample (n = 40) | Full Profile (Loci = 24) | Partial Profile | Mixed | Full + Minor | |||
|---|---|---|---|---|---|---|---|
| 1~10 | 11~19 | 20~23 | |||||
| Fingertip | 10 | 4 | 1 | 1 | 1 | 1 | 2 |
| Mobile phone | 10 | 7 | 1 | 1 | 0 | 0 | 1 |
| Urine | 10 | 9 | 0 | 0 | 0 | 0 | 1 |
| Gargle | 10 | 9 | 0 | 0 | 0 | 0 | 1 |
| Gargle | Urine | Fingertip |
|---|---|---|
| Streptococcus | Escherichia | Corynebacterium |
| Veillonella | Staphylococcus | Streptococcus |
| Prevotella | Finegoldia | Staphylococcus |
| Neisseria | Atopobium | Micrococcus |
| Haemophilus | Lactobacillus | Veillonella |
| Porphyromonas | Corynebacterium | Dermacoccus |
| Rothia | Gardnerella | Cutibacterium |
| Actinomyces | Campylobacter | Enhydrobacter |
| Campylobacter | Peptoniphilus | Sphingomonas |
| Tannerella | Anaerococcus | Lawsonella |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, S.; You, H.; Lee, S.; Lee, Y.; Kang, H.-G.; Sung, H.-J.; Choi, J.; Hyun, S. Individual Identification with Short Tandem Repeat Analysis and Collection of Secondary Information Using Microbiome Analysis. Genes 2022, 13, 85. https://doi.org/10.3390/genes13010085
Lee S, You H, Lee S, Lee Y, Kang H-G, Sung H-J, Choi J, Hyun S. Individual Identification with Short Tandem Repeat Analysis and Collection of Secondary Information Using Microbiome Analysis. Genes. 2022; 13(1):85. https://doi.org/10.3390/genes13010085
Chicago/Turabian StyleLee, Solip, Heesang You, Songhee Lee, Yeongju Lee, Hee-Gyoo Kang, Ho-Joong Sung, Jiwon Choi, and Sunghee Hyun. 2022. "Individual Identification with Short Tandem Repeat Analysis and Collection of Secondary Information Using Microbiome Analysis" Genes 13, no. 1: 85. https://doi.org/10.3390/genes13010085