
Spatial and Temporal Gene Function Studies in Rodents: Towards Gene-Based Therapies for Autism Spectrum Disorder



Abstract
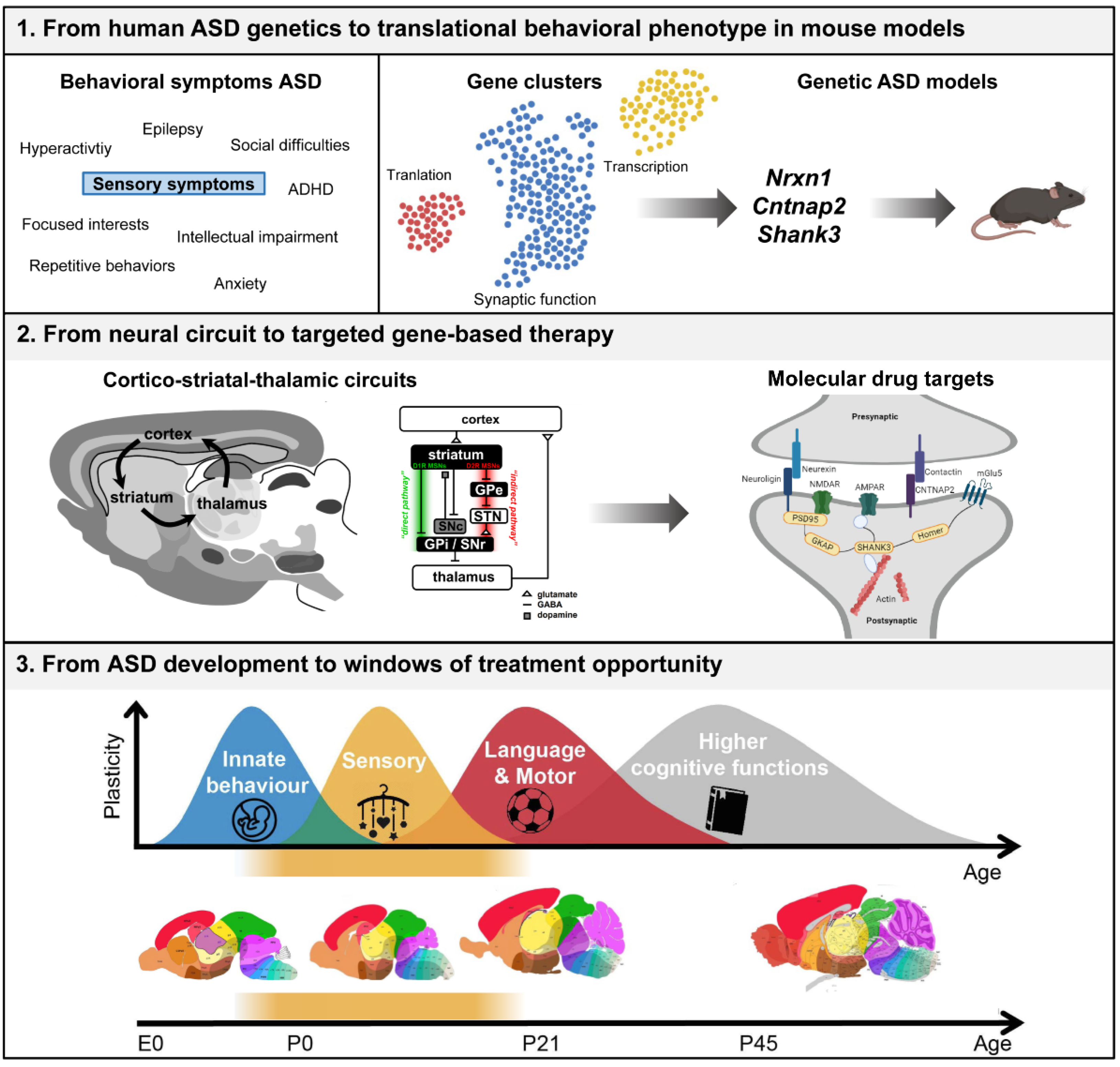
:1. Introduction
2. From Human ASD Genetics to Translational Behavioral Phenotype in Mouse Models
2.1. Behavioral Phenotypes in Genetic Mouse Models of Synaptic Dysfunction in ASD
2.2. Early Behavioral Phenotypes

{kind=link}
| Genetic ASD Rodent Model | Sensory Phenotypes | Social Phenotypes | Stereotyped and Repetitive Behaviors | Other Phenotypes | Early Behavioral Phenotypes |
|---|---|---|---|---|---|
| Nrxn1 | ↑ Response to startle [31,36] | ↑ Aggression [35,47] | ↑ Grooming [31] | ↑ Locomotion activity [36,38] | PND 2–12 ↓ Number and complexity of USVs [47,48] |
| ↓ Acoustic prepulse inhibition [31] | ↑ Preference for social stimulus versus nonsocial stimulus [35] | ↑ Novelty responsiveness and habituation [38] | ↑ Motor performance [31] | PND 5–15 ↓ Developmental milestones: body weight, length, vertical screen grasp negative geotaxis, ear canal opening and cliff avoidance [47] | |
| ↓ Eyeblink conditioning [48] | ↓ Social interaction [35,47] | ↓ Nest building [31,35] | ↑ Anxiety [35] | PND 26 ↑ Locomotion [48] | |
| ↓ Preference for social novelty [46] | ↓ Instrumental and spatial learning [36] | PND 27–30 ↓ Social interaction during social play [48] | |||
| ↓ Reward processing [49] | PND 30 ↑ Object investigation in novel object task [48] | ||||
| PND 30 ↓ Social investigative behavior [47] | |||||
| PND 30–35 ↓ Performance in food-reward task [48] | |||||
| PND 30–35 ↓ Prosocial helping behavior and nurturing behavior [48] | |||||
| Cntnap2 | ↑ Mechanical sensitivity in the von Frey tests [54] | ↓ Three chamber social preference [30,37] | ↑ Stereotypic motor movements and behavioral inflexibility [30] | ↓ Freezing [37] | PND 3–12 ↓ USVs [51] |
| ↑ Response in pain sensitivity to algogens [54] | ↓ Vocalizations in response to estrous females urine [37] | ↑ grooming and digging [30] | ↑ Epileptic seizures and epileptiform activity [30] | PND 4 ↑ Rolling on the side during walking [37] | |
| ↑ Acoustic startle responses and moderate-intensity sound avoidance [53] | ↑ locomotor activity and gaiting phenotype [30,37] | ↓ Morris water maze learning [30] | PND 4–15 ↑ Geotaxis [37] | ||
| ↑ Tactile–acoustic prepulse inhibition [39] | ↓ Nest building [30] | ↓ Spontaneous alternations [30] | PND 21 ↓ Social interaction in juvenile play [30] | ||
| ↑ Response to air-puff [39,59] | |||||
| ↑↓ Prepulse inhibition [37,53] | |||||
| ↓ Audio-visual temporal recalibration [53] | |||||
| ↓ Withdrawal latency in the hot plate test [54] | |||||
| ↓ Eyeblink conditioning, cerebellar sensory learning [52] | |||||
| Shank3 | ↑ Hotplate sensitivity [34] | ↓ Preference for social novelty [32,33,34,39,62,63] | ↑ Self-grooming [32,33,34,40,41,42,58,61,62,63] | ↑ Anxiety-like behavior [32,33,34,39] | PND 4 ↓ Ultrasonic vocalizations [58] |
| ↑ Tactile prepulse inhibition [39,59] | ↓ Preference for social stimulus versus nonsocial stimulus [32,33,39,40,62] | ↑ Circling behavior [34,40,41] | ↑ Dominance-like behavior [32] | PND 5–13 ↓ Negative geotaxis [41] | |
| ↑ Response to air puff [39,59] | ↓ Social interaction [40,58,64] | ↑ Repetitive object exploration [40] | ↓ Locomotor activity [32,34,41,58] | PND 10–12 ↓ Mid-air righting task [41] | |
| ↓ Eyeblink conditioning, cerebellar sensory learning) [52] | ↓ Adult USVs [40,58,64] | ↑ Repetitive hole board exploration [40,58] | ↓ Motor performance [32,34,40,41,42,58,63] | PND 12–15 ↓ Response auditory startle [41] | |
| ↓ Acoustic startle response [32,41] | ↓ Marble burying [34,41] | ↓ Object recognition and exploration [34,41,42] | PND 13, 14 ↓ Grasping reflex [41] | ||
| ↓ Acoustic prepulse inhibition [32] | ↓ Nest building [41] | ↓ Barnes maze training and reversal [41] | PND 14, 17–21 ↓ Weight [41] | ||
| ↓ Buried food test [41] | ↓ Contextual fear testing [41] | PND 15 ↓ Home cage nest preference [58] | |||
| ↓ Exploration of nonsocial odors [41] | ↓ Spatial learning in the Morris water maze [34,40,62] | PND 15–20 ↓ Wire suspension [41] | |||
| ↓ Texture recognition [39,59] | ↓ Striatal dependent learning [58] | PND 21–25 ↓ Social interaction [32,42] | |||
| ↓ Heat hyperalgesia in inflammatory and neuropathic pain [60] | ↓ T-maze reversal [62] | PND 42–56 ↓ Social interaction [61] |
3. From Neural Circuit to Targeted Gene-Based Therapy
3.1. Spatiotemporal Expression of the Synaptic Genes Nrnx1, Cntnap2, and Shank3
3.2. Molecular and Downstream Targets of Nrxn1, Cntnap2, and Shank3
4. From ASD Development to Windows of Treatment Opportunity
4.1. Timing of Intervention in ASD
4.2. Treatment of Tactile Sensory System
4.3. Treatment of Proteins Related to Synaptic Plasticity in ASD
4.4. Translation of Drug Target Findings from Rodents to Humans
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Fountain, C.; Winter, A.S.; Bearman, P.S. Six Developmental Trajectories Characterize Children with Autism. Pediatrics 2012, 129, e1112–e1120. [Google Scholar] [CrossRef] [Green Version]
- Kas, M.J.; Glennon, J.C.; Buitelaar, J.; Ey, E.; Biemans, B.; Crawley, J.; Ring, R.H.; Lajonchere, C.; Esclassan, F.; Talpos, J.; et al. Assessing behavioural and cognitive domains of autism spectrum disorders in rodents: Current status and future perspec-tives. Psychopharmacology 2014, 231, 1125–1146. [Google Scholar] [CrossRef] [Green Version]
- Estes, A.; Zwaigenbaum, L.; Gu, H.; John, T.S.; Paterson, S.; Elison, J.T.; Hazlett, H.; Botteron, K.; Dager, S.R.; Schultz, R.T.; et al. Behavioral, cognitive, and adaptive development in infants with autism spectrum disorder in the first 2 years of life. J. Neurodev. Disord. 2015, 7, 1–10. [Google Scholar] [CrossRef] [Green Version]
- McCormick, C.; Hepburn, S.; Young, G.S.; Rogers, S.J. Sensory symptoms in children with autism spectrum disorder, other developmental disorders and typical development: A longitudinal study. Autism 2016, 20, 572–579. [Google Scholar] [CrossRef] [Green Version]
- Hornix, B.E.; Havekes, R.; Kas, M.J.H. Multisensory cortical processing and dysfunction across the neuropsychiatric spec-trum. Neurosci. Biobehav. Rev. 2019, 97, 138–151. [Google Scholar] [CrossRef] [PubMed]
- American Psychiatric Association (APA). Diagnostic and Statistical Manual of Mental Disorders: DSM-5, 5th ed.; American Psychiatric Association: Washington, DC, USA, 2013; ISBN 978-0-89042-554-1. [Google Scholar]
- Tomchek, S.D.; Dunn, W. Sensory Processing in Children with and Without Autism: A Comparative Study Using the Short Sensory Profile. Am. J. Occup. Ther. 2007, 61, 190–200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tavassoli, T.; Miller, L.J.; Schoen, A.S.; Nielsen, D.M.; Baron-Cohen, S. Sensory over-responsivity in adults with autism spectrum conditions. Autism 2014, 18, 428–432. [Google Scholar] [CrossRef]
- Hilton Jacquelyn, C.L.; Harper, J.D.; Kueker, R.H.; Lang, A.R.; Abbacchi, A.M.; Todorov, A.; LaVesser, P.D. Sensory Responsiveness as a Predictor of Social Severity in Children with High Functioning Autism Spectrum Disorders. J. Autism Dev. Disord. 2010, 40, 937–945. [Google Scholar] [CrossRef]
- Schulz, E.S.; Stevenson, A.R. Sensory hypersensitivity predicts repetitive behaviours in autistic and typically-developing children. Autism 2019, 23, 1028–1041. [Google Scholar] [CrossRef]
- Bailey, A.; Le Couteur, A.; Gottesman, I.; Bolton, P.; Simonoff, E.; Yuzda, E.; Rutter, M.L. Autism as a strongly genetic disorder: Evidence from a British twin study. Psychol. Med. 1995, 25, 63–77. [Google Scholar] [CrossRef] [PubMed]
- Le Couteur, A.; Bailey, A.; Goode, S.; Pickles, A.; Gottesman, I.; Robertson, S.; Rutter, M. A Broader Phenotype of Autism: The Clinical Spectrum in Twins. J. Child Psychol. Psychiatry 1996, 37, 785–801. [Google Scholar] [CrossRef]
- Liu, X.; Takumi, T. Genomic and genetic aspects of autism spectrum disorder. Biochem. Biophys. Res. Commun. 2014, 452, 244–253. [Google Scholar] [CrossRef] [Green Version]
- Tick, B.; Bolton, P.; Happé, F.; Rutter, M.L.; Rijsdijk, F. Heritability of autism spectrum disorders: A meta-analysis of twin studies. J. Child Psychol. Psychiatry 2016, 57, 585–595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sandin, S.; Lichtenstein, P.; Kuja-Halkola, R.; Larsson, H.; Hultman, C.M.; Reichenberg, A. The Familial Risk of Autism. J. Am. Med. Assoc. 2014, 311, 1770–1777. [Google Scholar] [CrossRef] [PubMed]
- Sfari, G. Gene. Available online: https://www.sfari.org/resource/sfari-gene/ (accessed on 16 November 2021).
- Ramaswami, G.; Geschwind, D.H. Genetics of autism spectrum disorder. In Handbook of Clinical Neurology; University of California: Los Angeles, CA, USA, 2018; Volume 147. [Google Scholar]
- Abrahams, B.S.; Arking, E.D.; Campbell, D.B.; Mefford, H.C.; Morrow, E.M.; Weiss, A.L.; Menashe, I.; Wadkins, T.; Banerjee-Basu, S.; Packer, A. SFARI Gene 2.0: A community-driven knowledgebase for the autism spectrum disorders (ASDs). Mol. Autism 2013, 4, 36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moyses-Oliveira, M.; Yadav, R.; Erdin, S.; Talkowski, E.M. New gene discoveries highlight functional convergence in autism and related neurodevelopmental disorders. Curr. Opin. Genet. Dev. 2020, 65, 195–206. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, A.; Zhang, R.; Yao, V.; Theesfeld, C.L.; Wong, A.K.; Tadych, A.; Volfovsky, N.; Packer, A.; Lash, A.; Troyanskaya, O.G. Genome-wide prediction and functional characterization of the genetic basis of autism spectrum disorder. Nat. Neurosci. 2016, 19, 1454–1462. [Google Scholar] [CrossRef] [Green Version]
- Baranova, J.; Dragunas, G.; Botellho, M.C.S.; Ayub, A.L.P.; Bueno-Alves, R.; Alencar, R.R.; Papaiz, D.D.; Sogayar, M.C.; Ulrich, H.; Correa, R.G. Autism Spectrum Disorder: Signaling Pathways and Prospective Therapeutic Targets. Cell. Mol. Neurobiol. 2021, 41, 619–649. [Google Scholar] [CrossRef]
- Sacca, R.; Engle, S.J.; Qin, W.; Stock, J.L.; McNeish, J.D. Genetically Engineered Mouse Models in Drug Discovery Research. Methods Mol. Biol. 2010, 602, 37–54. [Google Scholar] [CrossRef] [PubMed]
- Bellosta, P.; Soldano, A. Dissecting the Genetics of Autism Spectrum Disorders: A Drosophila Perspective. Front. Physiol. 2019, 10, 987. [Google Scholar] [CrossRef]
- Owald, D.; Khorramshahi, O.; Gupta, V.K.; Banovic, D.; Depner, H.; Fouquet, W.; Wichmann, C.; Mertel, S.; Eimer, S.; Reynolds, E.; et al. Cooperation of Syd-1 with Neurexin synchronizes pre- with postsynaptic assembly. Nat. Neurosci. 2012, 15, 1219–1226. [Google Scholar] [CrossRef]
- Anderson, G.R.; Aoto, J.; Tabuchi, K.; Földy, C.; Covy, J.; Yee, A.X.; Wu, D.; Lee, S.-J.; Chen, L.; Malenka, R.C.; et al. β-Neurexins Control Neural Circuits by Regulating Synaptic Endocannabinoid Signaling. Cell 2015, 162, 593–606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Missler, M.; Zhang, W.; Rohlmann, A.; Kattenstroth, G.; Hammer, R.E.; Gottmann, K.; Südhof, T.C. α-Neurexins couple Ca2+ channels to synaptic vesicle exocytosis. Nat. Cell Biol. 2003, 423, 939–948. [Google Scholar] [CrossRef] [PubMed]
- Vecchia, E.D.; Mortimer, N.; Palladino, V.S.; Kittel-Schneider, S.; Lesch, K.-P.; Reif, A.; Schenck, A.; Norton, W.H. Cross-species models of attention-deficit/hyperactivity disorder and autism spectrum disorder: Lessons from CNTNAP2, AD-GRL3, and PARK2. Psychiatr. Genet. 2019, 29, 1–17. [Google Scholar] [CrossRef]
- Silverman, J.L.; Yang, M.; Lord, C.; Crawley, J.N. Behavioural phenotyping assays for mouse models of autism. Nat. Rev. Neurosci. 2010, 11, 490–502. [Google Scholar] [CrossRef] [Green Version]
- Balasco, L.; Provenzano, G.; Bozzi, Y. Sensory Abnormalities in Autism Spectrum Disorders: A Focus on the Tactile Domain, From Genetic Mouse Models to the Clinic. Front. Psychiatry 2020, 10, 1016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peñagarikano, O.; Abrahams, B.S.; Herman, E.I.; Winden, K.D.; Gdalyahu, A.; Dong, H.; Sonnenblick, L.I.; Gruver, R.; Almajano, J.; Bragin, A.; et al. Absence of CNTNAP2 Leads to Epilepsy, Neuronal Migration Abnormalities, and Core Autism-Related Deficits. Cell 2011, 147, 235–246. [Google Scholar] [CrossRef] [Green Version]
- Etherton, M.R.; Blaiss, C.A.; Powell, C.M.; Sudhof, T.C. Mouse neurexin-1 deletion causes correlated electrophysiological and behavioral changes consistent with cognitive impairments. Proc. Natl. Acad. Sci. USA 2009, 106, 17998–18003. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Kaiser, T.; Monteiro, P.; Zhang, X.; Van der Goes, M.S.; Wang, D.; Barak, B.; Zeng, M.; Li, C.; Lu, C.; et al. Mice with Shank3 Mutations Associated with ASD and Schizophrenia Display Both Shared and Distinct Defects. Neuron 2016, 89, 147–162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peça, J.; Feliciano, C.; Ting, J.; Wang, W.; Wells, M.F.; Venkatraman, T.N.; Lascola, C.D.; Fu, Z.; Feng, G. Shank3 mutant mice display autistic-like behaviours and striatal dysfunction. Nat. Cell Biol. 2011, 472, 437–442. [Google Scholar] [CrossRef] [Green Version]
- Kouser, M.; Speed, H.; Dewey, C.M.; Reimers, J.M.; Widman, A.J.; Gupta, N.; Liu, S.; Jaramillo, T.C.; Bangash, M.; Xiao, B.; et al. Loss of Predominant Shank3 Isoforms Results in Hippocampus-Dependent Impairments in Behavior and Synaptic Transmission. J. Neurosci. 2013, 33, 18448–18468. [Google Scholar] [CrossRef]
- Grayton, H.M.; Missler, M.; Collier, D.A.; Fernandes, C. Altered Social Behaviours in Neurexin 1α Knockout Mice Resemble Core Symptoms in Neurodevelopmental Disorders. PLoS ONE 2013, 8, e67114. [Google Scholar] [CrossRef] [Green Version]
- Esclassan, F.; Francois, J.; Phillips, K.G.; Loomis, S.; Gilmour, G. Phenotypic characterization of nonsocial behavioral impairment in neurexin 1α knockout rats. Behav. Neurosci. 2015, 129, 74–85. [Google Scholar] [CrossRef] [PubMed]
- Brunner, D.; Kabitzke, P.; He, D.; Cox, K.; Thiede, L.; Hanania, T.; Sabath, E.; Alexandrov, V.; Saxe, M.; Peles, E.; et al. Comprehensive Analysis of the 16p11.2 Deletion and Null Cntnap2 Mouse Models of Autism Spectrum Disorder. PLoS ONE 2015, 10, e0134572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laarakker, M.C.; Reinders, N.R.; Bruining, H.; Ophoff, R.A.; Kas, M.J.H. Sex-Dependent Novelty Response in Neurexin-1α Mutant Mice. PLoS ONE 2012, 7, e31503. [Google Scholar] [CrossRef] [Green Version]
- Orefice, L.L.; Mosko, J.R.; Morency, D.; Wells, M.F.; Tasnim, A.; Mozeika, S.M.; Ye, M.; Chirila, A.M.; Emanuel, A.; Rankin, G.; et al. Targeting Peripheral Somatosensory Neurons to Improve Tactile-Related Phenotypes in ASD Models. Cell 2019, 178, 867–886.e24. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; McCoy, P.A.; Rodriguiz, R.M.; Pan, Y.; Je, H.S.; Roberts, A.C.; Kim, C.J.; Berrios, J.; Colvin, J.S.; Bousquet-Moore, D.; et al. Synaptic dysfunction and abnormal behaviors in mice lacking major isoforms of Shank. Hum. Mol. Genet. 2011, 20, 3093–3108. [Google Scholar] [CrossRef] [Green Version]
- Drapeau, E.; Dorr, N.P.; Elder, G.A.; Buxbaum, J.D. Absence of strong strain effects in behavioral analyses of Shank3-deficient mice. Dis. Model. Mech. 2014, 7, 667–681. [Google Scholar] [CrossRef] [Green Version]
- Yang, M.; Bozdagi, O.; Scattoni, M.L.; Wöhr, M.; Roullet, F.I.; Katz, A.M.; Abrams, D.N.; Kalikhman, D.; Simon, H.; Woldeyohannes, L.; et al. Reduced Excitatory Neurotransmission and Mild Autism-Relevant Phenotypes in Adolescent Shank3 Null Mutant Mice. J. Neurosci. 2012, 32, 6525–6541. [Google Scholar] [CrossRef]
- Ching, M.; Shen, Y.; Tan, W.-H.; Jeste, S.; Morrow, E.; Chen, X.; Mukaddes, N.M.; Yoo, S.-Y.; Hanson, E.; Hundley, R.; et al. Deletions of NRXN1 (neurexin-1) predispose to a wide spectrum of developmental disorders. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 2010, 153B, 937–947. [Google Scholar] [CrossRef]
- Reissner, C.; Runkel, F.; Missler, M. Neurexins. Genome Biol. 2013, 14, 1–15. [Google Scholar] [CrossRef]
- Hu, Z.; Xiao, X.; Zhang, Z.; Li, M. Genetic insights and neurobiological implications from NRXN1 in neuropsychiatric dis-orders. Mol. Psychiatry 2019, 24, 1400–1414. [Google Scholar] [CrossRef]
- Dachtler, J.; Ivorra, J.L.; Rowland, T.E.; Lever, C.; Rodgers, R.J.; Clapcote, S.J. Heterozygous deletion of α-neurexin I or α-neurexin II results in behaviors relevant to autism and schizophrenia. Behav. Neurosci. 2015, 129, 765–776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armstrong, E.C.; Caruso, A.; Servadio, M.; Andreae, L.; Trezza, V.; Scattoni, M.L.; Fernandes, C. Assessing the developmental trajectory of mouse models of neurodevelopmental disorders: Social and communication deficits in mice with Neurexin 1α deletion. Genes Brain Behav. 2020, 19, e12630. [Google Scholar] [CrossRef] [PubMed]
- Kight, K.E.; Argue, K.J.; Bumgardner, J.G.; Bardhi, K.; Waddell, J.; McCarthy, M.M. Social behavior in prepubertal neurexin 1α deficient rats: A model of neurodevelopmental disorders. Behav. Neurosci. 2021, 135, 782–803. [Google Scholar] [CrossRef] [PubMed]
- Alabi, O.O.; Davatolhagh, M.F.; Robinson, M.; Fortunato, M.P.; Cifuentes, L.V.; Kable, J.W.; Fuccillo, M.V. Disruption of Nrxn1α within excitatory forebrain circuits drives value-based dysfunction. eLife 2020, 9, e54838. [Google Scholar] [CrossRef]
- Lu, Z.; Reddy, M.V.V.V.S.; Liu, J.; Kalichava, A.; Zhang, L.; Chen, F.; Wang, Y.; Holthauzen, L.M.F.; White, M.A.; Seshadrinathan, S.; et al. Molecular Architecture of Contactin-associated Protein-like 2 (CNTNAP2) and Its Interaction with Contactin 2 (CNTN2). J. Biol. Chem. 2016, 291, 24133–24147. [Google Scholar] [CrossRef] [Green Version]
- Scattoni, M.L.; Crawley, J.; Ricceri, L. Ultrasonic vocalizations: A tool for behavioural phenotyping of mouse models of neurodevelopmental disorders. Neurosci. Biobehav. Rev. 2009, 33, 508–515. [Google Scholar] [CrossRef] [Green Version]
- Kloth, A.D.; Badura, A.; Li, A.; Cherskov, A.; Connolly, S.G.; Giovannucci, A.; Bangash, M.A.; Grasselli, G.; Peñagarikano, O.; Piochon, C.; et al. Cerebellar associative sensory learning defects in five mouse autism models. eLife 2015, 4, 4. [Google Scholar] [CrossRef]
- Scott, K.E.; Kazazian, K.; Mann, R.S.; Möhrle, D.; Schormans, A.L.; Schmid, S.; Allman, B.L. Loss of Cntnap2 in the Rat Causes Autism-Related Alterations in Social Interactions, Stereotypic Behavior, and Sensory Processing. Autism Res. 2020, 13, 1698–1717. [Google Scholar] [CrossRef]
- Dawes, J.M.; Weir, G.; Middleton, S.J.; Patel, R.; Chisholm, K.; Pettingill, P.; Peck, L.; Sheridan, J.; Shakir, A.; Jacobson, L.; et al. Immune or Genetic-Mediated Disruption of CASPR2 Causes Pain Hypersensitivity Due to Enhanced Primary Afferent Excitability. Neuron 2018, 97, 806–822.e10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gordon, A.; Salomon, D.; Barak, N.; Pen, Y.; Tsoory, M.; Kimchi, T.; Peles, E. Expression of Cntnap2 (Caspr2) in multiple levels of sensory systems. Mol. Cell. Neurosci. 2016, 70, 42–53. [Google Scholar] [CrossRef] [PubMed]
- Monteiro, P.; Feng, G. Shank proteins: Roles at the synapse and in autism spectrum disorder. Nat. Rev. Neurosci. 2017, 18, 147–157. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.-H.; Ehlers, M.D. Modeling Autism by SHANK Gene Mutations in Mice. Neuron 2013, 78, 8–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Bey, A.L.; Katz, B.M.; Badea, A.; Kim, N.; David, L.K.; Duffney, L.J.; Kumar, S.; Mague, S.D.; Hulbert, S.; et al. Altered mGluR5-Homer scaffolds and corticostriatal connectivity in a Shank3 complete knockout model of autism. Nat. Commun. 2016, 7, 11459. [Google Scholar] [CrossRef] [Green Version]
- Orefice, L.L.; Zimmerman, A.L.; Chirila, A.M.; Sleboda, S.J.; Head, J.P.; Ginty, D.D. Peripheral Mechanosensory Neuron Dysfunction Underlies Tactile and Behavioral Deficits in Mouse Models of ASDs. Cell 2016, 166, 299–313. [Google Scholar] [CrossRef] [Green Version]
- Han, Q.; Kim, Y.H.; Wang, X.; Liu, D.; Zhang, Z.-J.; Bey, A.L.; Lay, M.; Chang, W.; Berta, T.; Zhang, Y.; et al. SHANK3 Deficiency Impairs Heat Hyperalgesia and TRPV1 Signaling in Primary Sensory Neurons. Neuron 2016, 92, 1279–1293. [Google Scholar] [CrossRef] [Green Version]
- Duffney, L.J.; Zhong, P.; Wei, J.; Matas, E.; Cheng, J.; Qin, L.; Ma, K.; Dietz, D.; Kajiwara, Y.; Buxbaum, J.; et al. Autism-like Deficits in Shank3-Deficient Mice Are Rescued by Targeting Actin Regulators. Cell Rep. 2015, 11, 1400–1413. [Google Scholar] [CrossRef] [Green Version]
- Vicidomini, C.; Ponzoni, L.; Lim, D.; Schmeisser, M.; Reim, D.; Morello, N.; Orelanna, D.; Tozzi, A.; Durante, V.; Scalmani, P.; et al. Pharmacological enhancement of mGlu5 receptors rescues behavioral deficits in SHANK3 knock-out mice. Mol. Psychiatry 2016, 22, 689–702. [Google Scholar] [CrossRef] [Green Version]
- Matas, E.; Maisterrena, A.; Thabault, M.; Balado, E.; Francheteau, M.; Balbous, A.; Galvan, L.; Jaber, M. Major motor and gait deficits with sexual dimorphism in a Shank3 mutant mouse model. Mol. Autism 2021, 12, 1–16. [Google Scholar] [CrossRef]
- Bozdagi, O.; Sakurai, T.; Papapetrou, D.; Wang, X.; Dickstein, D.L.; Takahashi, N.; Kajiwara, Y.; Yang, M.; Katz, A.M.; Scattoni, M.L.; et al. Haploinsufficiency of the autism-associated Shank3 gene leads to deficits in synaptic function, social interaction, and social communication. Mol. Autism 2010, 1, 15. [Google Scholar] [CrossRef] [Green Version]
- Jenkins, A.K.; Paterson, C.; Wang, Y.; Hyde, T.M.; Kleinman, J.E.; Law, A.J. Neurexin 1 (NRXN1) splice isoform expression during human neocortical development and aging. Mol. Psychiatry 2016, 21, 701–706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harkin, L.F.; Lindsay, S.J.; Xu, Y.; Alzu’Bi, A.; Ferrara, A.; Gullon, E.A.; James, O.G.; Clowry, G.J. Neurexins 1–3 Each Have a Distinct Pattern of Expression in the Early Developing Human Cerebral Cortex. Cereb. Cortex 2016, 27, 216–232. [Google Scholar] [CrossRef] [PubMed]
- Fuccillo, M.V.; Földy, C.; Gokce, O.; Rothwell, P.; Sun, G.L.; Malenka, R.C.; Südhof, T.C. Single-Cell mRNA Profiling Reveals Cell-Type-Specific Expression of Neurexin Isoforms. Neuron 2015, 87, 326–340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davatolhagh, M.F.; Fuccillo, M.V. Neurexin1⍺ differentially regulates synaptic efficacy within striatal circuits. Cell Rep. 2021, 34, 108773. [Google Scholar] [CrossRef]
- Rodenas-Cuadrado, P.; Ho, J.; Vernes, S.C. Shining a light on CNTNAP2: Complex functions to complex disorders. Eur. J. Hum. Genet. 2014, 22, 171–178. [Google Scholar] [CrossRef]
- Abrahams, B.S.; Tentler, D.; Perederiy, J.V.; Oldham, M.C.; Coppola, G.; Geschwind, D.H. Genome-wide analyses of human perisylvian cerebral cortical patterning. Proc. Natl. Acad. Sci. USA 2007, 104, 17849–17854. [Google Scholar] [CrossRef] [Green Version]
- Poliak, S.; Gollan, L.; Martinez, R.; Custer, A.; Einheber, S.; Salzer, J.; Trimmer, J.S.; Shrager, P.; Peles, E. Caspr2, a New Member of the Neurexin Superfamily, Is Localized at the Juxtaparanodes of Myelinated Axons and Associates with K+ Channels. Neuron 1999, 24, 1037–1047. [Google Scholar] [CrossRef] [Green Version]
- Alarcón, M.; Abrahams, B.S.; Stone, J.L.; Duvall, J.A.; Perederiy, J.V.; Bomar, J.M.; Sebat, J.; Wigler, M.; Martin, C.L.; Ledbetter, D.H.; et al. Linkage, Association, and Gene-Expression Analyses Identify CNTNAP2 as an Autism-Susceptibility Gene. Am. J. Hum. Genet. 2008, 82, 150–159. [Google Scholar] [CrossRef] [Green Version]
- Böckers, T.M.; Mameza, M.G.; Kreutz, M.R.; Bockmann, J.; Weise, C.; Buck, F.; Richter, D.; Gundelfinger, E.D.; Kreienkamp, H.-J. Synaptic Scaffolding Proteins in Rat Brain: Ankyrin repeats of the multidomain Shank protein family interact with the cytoskeletal protein α-fodrin. J. Biol. Chem. 2001, 276, 40104–40112. [Google Scholar] [CrossRef] [Green Version]
- Ziats, C.A.; Grosvenor, L.P.; Sarasua, S.M.; Thurm, A.E.; Swedo, S.E.; Mahfouz, A.; Rennert, O.M.; Ziats, M.N. Functional genomics analysis of Phelan-McDermid syndrome 22q13 region during human neurodevelopment. PLoS ONE 2019, 14, e0213921. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Xu, Q.; Bey, A.L.; Lee, Y.; Jiang, Y.-H. Transcriptional and functional complexity of Shank3 provides a molecular framework to understand the phenotypic heterogeneity of SHANK3 causing autism and Shank3 mutant mice. Mol. Autism 2014, 5, 30. [Google Scholar] [CrossRef] [Green Version]
- Bey, A.L.; Wang, X.; Yan, H.; Kim, N.; Passman, R.L.; Yang, Y.; Cao, X.; Towers, A.J.; Hulbert, S.W.; Duffney, L.J.; et al. Brain region-specific disruption of Shank3 in mice reveals a dissociation for cortical and striatal circuits in autism-related behaviors. Transl. Psychiatry 2018, 8, 1–17. [Google Scholar] [CrossRef]
- Raab, M.; Boeckers, T.; Neuhuber, W. Proline-rich synapse-associated protein-1 and 2 (ProSAP1/Shank2 and ProSAP2/Shank3)—Scaffolding proteins are also present in postsynaptic specializations of the peripheral nervous system. Neuroscience 2010, 171, 421–433. [Google Scholar] [CrossRef]
- Zerbi, V.; Pagani, M.; Markicevic, M.; Matteoli, M.; Pozzi, D.; Fagiolini, M.; Bozzi, Y.; Galbusera, A.; Scattoni, M.L.; Provenzano, G.; et al. Brain mapping across 16 autism mouse models reveals a spectrum of functional connectivity subtypes. Mol. Psychiatry 2021, 1–11. [Google Scholar] [CrossRef]
- Fuccillo, M.V. Striatal Circuits as a Common Node for Autism Pathophysiology. Front. Neurosci. 2016, 10, 27. [Google Scholar] [CrossRef] [Green Version]
- Pak, C.; Danko, T.; Zhang, Y.; Aoto, J.; Anderson, G.; Maxeiner, S.; Yi, F.; Wernig, M.; Südhof, T.C. Human Neuropsychiatric Disease Modeling using Conditional Deletion Reveals Synaptic Transmission Defects Caused by Heterozygous Mutations in NRXN. Cell Stem Cell 2015, 17, 316–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, J.; Aoto, J.; Südhof, T.C. Alternative Splicing of Presynaptic Neurexins Differentially Controls Postsynaptic NMDA and AMPA Receptor Responses. Neuron 2019, 102, 993–1008.e5. [Google Scholar] [CrossRef]
- Anderson, G.R.; Galfin, T.; Xu, W.; Aoto, J.; Malenka, R.C.; Sudhof, T.C. Candidate autism gene screen identifies critical role for cell-adhesion molecule CASPR2 in dendritic arborization and spine development. Proc. Natl. Acad. Sci. USA 2012, 109, 18120–18125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gdalyahu, A.; Lázaro, M.; Penagarikano, O.; Golshani, P.; Trachtenberg, J.T.; Gescwind, D.H. The Autism Related Protein Contactin-Associated Protein-Like 2 (CNTNAP2) Stabilizes New Spines: An In Vivo Mouse Study. PLoS ONE 2015, 10, e0125633. [Google Scholar] [CrossRef]
- Varea, O.; Martin-De-Saavedra, M.D.; Kopeikina, K.J.; Schürmann, B.; Fleming, H.J.; Fawcett-Patel, J.M.; Bach, A.; Jang, S.; Peles, E.; Kim, E.; et al. Synaptic abnormalities and cytoplasmic glutamate receptor aggregates in contactin associated protein-like 2/Caspr2 knockout neurons. Proc. Natl. Acad. Sci. USA 2015, 112, 6176–6181. [Google Scholar] [CrossRef] [Green Version]
- Xing, X.; Zhang, J.; Wu, K.; Cao, B.; Li, X.; Jiang, F.; Hu, Z.; Xia, K.; Li, J.-D. Suppression of Akt-mTOR pathway rescued the social behavior in Cntnap2-deficient mice. Sci. Rep. 2019, 9, 3041. [Google Scholar] [CrossRef]
- Ehninger, D.; Silva, A.J. Rapamycin for treating Tuberous sclerosis and Autism spectrum disorders. Trends Mol. Med. 2011, 17, 78–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horresh, I.; Bar, V.; Kissil, J.L.; Peles, E. Organization of Myelinated Axons by Caspr and Caspr2 Requires the Cytoskeletal Adapter Protein 4.1B. J. Neurosci. 2010, 30, 2480–2489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horresh, I.; Poliak, S.; Grant, S.; Bredt, D.; Rasband, M.N.; Peles, E. Multiple Molecular Interactions Determine the Clustering of Caspr2 and Kv1 Channels in Myelinated Axons. J. Neurosci. 2008, 28, 14213–14222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mei, Y.; Monteiro, P.; Zhou, Y.; Kim, J.-A.; Gao, X.; Fu, Z.; Feng, G. Adult restoration of Shank3 expression rescues selective autistic-like phenotypes. Nat. Cell Biol. 2016, 530, 481–484. [Google Scholar] [CrossRef] [Green Version]
- Moutin, E.; Sakkaki, S.; Compan, V.; Bouquier, N.; Giona, F.; Areias, J.; Goyet, E.; Hemonnot-Girard, A.-L.; Seube, V.; Glasson, B.; et al. Restoring glutamate receptosome dynamics at synapses rescues autism-like deficits in Shank3-deficient mice. Mol. Psychiatry 2021, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Peixoto, R.T.; Wang, W.; Croney, D.M.; Kozorovitskiy, Y.; Sabatini, B.L. Early hyperactivity and precocious maturation of corticostriatal circuits in Shank3B−/− mice. Nat. Neurosci. 2016, 19, 716–724. [Google Scholar] [CrossRef]
- Platt, R.J.; Zhou, Y.; Slaymaker, I.M.; Shetty, A.; Weisbach, N.R.; Kim, J.-A.; Sharma, J.; Desai, M.; Sood, S.; Kempton, H.R.; et al. Chd8 Mutation Leads to Autistic-like Behaviors and Impaired Striatal Circuits. Cell Rep. 2017, 19, 335–350. [Google Scholar] [CrossRef] [Green Version]
- Kweon, H.; Jung, W.B.; Im, G.H.; Ryoo, J.; Lee, J.-H.; Do, H.; Choi, Y.; Song, Y.-H.; Jung, H.; Park, H.; et al. Excitatory neuronal CHD8 in the regulation of neocortical development and sensory-motor behaviors. Cell Rep. 2021, 34, 108780. [Google Scholar] [CrossRef]
- Li, W.; Pozzo-Miller, L. Dysfunction of the corticostriatal pathway in autism spectrum disorders. J. Neurosci. Res. 2020, 98, 2130–2147. [Google Scholar] [CrossRef]
- Varghese, M.; Keshav, N.; Jacot-Descombes, S.; Warda, T.; Wicinski, B.; Dickstein, D.L.; Harony-Nicolas, H.; De Rubeis, S.; Drapeau, E.; Buxbaum, J.; et al. Autism spectrum disorder: Neuropathology and animal models. Acta Neuropathol. 2017, 134, 537–566. [Google Scholar] [CrossRef]
- Hoffmann, A.; Spengler, D. Chromatin Remodeler CHD8 in Autism and Brain Development. J. Clin. Med. 2021, 10, 366. [Google Scholar] [CrossRef]
- Smith, E.S.; Smith, D.R.; Eyring, C.; Braileanu, M.; Smith-Connor, K.S.; Tan, Y.E.; Fowler, A.Y.; Hoffman, G.E.; Johnston, M.V.; Kannan, S.; et al. Altered trajectories of neurodevelopment and behavior in mouse models of Rett syndrome. Neurobiol. Learn. Mem. 2019, 165, 106962. [Google Scholar] [CrossRef]
- Jaramillo, T.C.; Xuan, Z.; Reimers, J.M.; Escamilla, C.O.; Liu, S.; Powell, C.M. Early Restoration of Shank3 Expression in Shank3 Knock-Out Mice Prevents Core ASD-Like Behavioral Phenotypes. Eneuro 2020, 7. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Huang, C.-C.; Dai, Y.; Luo, Q.; Ji, Y.; Wang, K.; Deng, S.; Yu, J.; Xu, M.; Du, X.; et al. Symptom improvement in children with autism spectrum disorder following bumetanide administration is associated with decreased GABA/glutamate ratios. Transl. Psychiatry 2020, 10, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Sprengers, J.J.; van Andel, D.M.; Zuithoff, N.P.; Keijzer-Veen, M.G.; Schulp, A.J.; Scheepers, F.E.; Lilien, M.R.; Oranje, B.; Bruining, H. Bumetanide for Core Symptoms of Autism Spectrum Disorder (BAMBI): A Single Center, Double-Blinded, Participant-Randomized, Placebo-Controlled, Phase-2 Superiority Trial. J. Am. Acad. Child Adolesc. Psychiatry 2021, 60, 865–876. [Google Scholar] [CrossRef]
- Bruining, H.; Hardstone, R.; Juarez-Martinez, E.L.; Sprengers, J.; Avramiea, A.-E.; Simpraga, S.; Houtman, S.J.; Poil, S.-S.; Dallares, E.; Palva, S.; et al. Measurement of excitation-inhibition ratio in autism spectrum disorder using critical brain dynamics. Sci. Rep. 2020, 10, 9195. [Google Scholar] [CrossRef] [PubMed]
- Schaffler, M.D.; Middleton, L.J.; Abdus-Saboor, I. Mechanisms of Tactile Sensory Phenotypes in Autism: Current Under-standing and Future Directions for Research. Curr. Psychiatry Rep. 2019, 21, 134. [Google Scholar] [CrossRef] [Green Version]
- McCoy, E.S.; Taylor-Blake, B.; Aita, M.; Simon, J.M.; Philpot, B.D.; Zylka, M.J. Enhanced Nociception in Angelman Syndrome Model Mice. J. Neurosci. 2017, 37, 10230–10239. [Google Scholar] [CrossRef] [PubMed]
- Tang, G.; Gudsnuk, K.; Kuo, S.-H.; Cotrina, M.L.; Rosoklija, G.; Sosunov, A.; Sonders, M.S.; Kanter, E.; Castagna, C.; Yamamoto, A.; et al. Loss of mTOR-Dependent Macroautophagy Causes Autistic-like Synaptic Pruning Deficits. Neuron 2014, 83, 1131–1143. [Google Scholar] [CrossRef] [Green Version]
- Xing, X.; Wu, K.; Dong, Y.; Zhou, Y.; Zhang, J.; Jiang, F.; Hu, W.-P.; Li, J.-D. Hyperactive Akt-mTOR pathway as a therapeutic target for pain hypersensitivity in Cntnap2-deficient mice. Neuropharmacology 2020, 165, 107816. [Google Scholar] [CrossRef] [PubMed]
- Mizuguchi, M.; Ikeda, H.; Kagitani-Shimono, K.; Yoshinaga, H.; Suzuki, Y.; Aoki, M.; Endo, M.; Yonemura, M.; Kubota, M. Everolimus for epilepsy and autism spectrum disorder in tuberous sclerosis complex: EXIST-3 substudy in Japan. Brain Dev. 2019, 41, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Overwater, I.E.; Rietman, A.B.; Mous, S.E.; Heus, K.B.-D.; Rizopoulos, D.; Hoopen, L.W.T.; Van Der Vaart, T.; Jansen, F.E.; Elgersma, Y.; Moll, H.A.; et al. A randomized controlled trial with everolimus for IQ and autism in tuberous sclerosis complex. Neurology 2019, 93, e200–e209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Posar, A.; Visconti, P. Autism spectrum disorder and mammalian target of rapamycin system. J. Pediatr. Neurosci. 2020, 15, 164–165. [Google Scholar] [CrossRef]
- Amegandjin, C.A.; Choudhury, M.; Jadhav, V.; Carriço, J.N.; Quintal, A.; Berryer, M.; Snapyan, M.; Chattopadhyaya, B.; Saghatelyan, A.; Di Cristo, G. Sensitive period for rescuing parvalbumin interneurons connectivity and social behavior deficits caused by TSC1 loss. Nat. Commun. 2021, 12, 3653. [Google Scholar] [CrossRef]
- Vogt, D.; Cho, K.; Shelton, S.M.; Paul, A.; Huang, Z.J.; Sohal, V.S.; Rubenstein, J.L.R. MouseCntnap2and HumanCNTNAP2ASD Alleles Cell Autonomously Regulate PV+ Cortical Interneurons. Cereb. Cortex 2017, 28, 3868–3879. [Google Scholar] [CrossRef] [Green Version]
- Zikopoulos, B.; Barbas, H. Altered neural connectivity in excitatory and inhibitory cortical circuits in autism. Front. Hum. Neurosci. 2013, 7, 609. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Li, C.; Chen, Q.; Van Der Goes, M.-S.; Hawrot, J.; Yao, A.Y.; Gao, X.; Lu, C.; Zang, Y.; Zhang, Q.; et al. Striatopallidal dysfunction underlies repetitive behavior in Shank3-deficient model of autism. J. Clin. Investig. 2017, 127, 1978–1990. [Google Scholar] [CrossRef] [Green Version]
- Borovac, J.; Bosch, M.; Okamoto, K. Regulation of actin dynamics during structural plasticity of dendritic spines: Signaling messengers and actin-binding proteins. Mol. Cell. Neurosci. 2018, 91, 122–130. [Google Scholar] [CrossRef]
- de Jong, J.O.; Llapashtica, C.; Genestine, M.; Strauss, K.; Provenzano, F.; Sun, Y.; Zhu, H.; Cortese, G.P.; Brundu, F.; Brigatti, K.W.; et al. Cortical overgrowth in a preclinical forebrain organoid model of CNTNAP2-associated autism spectrum disorder. Nat. Commun. 2021, 12, 4087. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Bhattacharyya, A. Human Models Are Needed for Studying Human Neurodevelopmental Disorders. Am. J. Hum. Genet. 2018, 103, 829–857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Urresti, J.; Zhang, P.; Moran-Losada, P.; Yu, N.-K.; Negraes, P.D.; Trujillo, C.A.; Antaki, D.; Amar, M.; Chau, K.; Pramod, A.B.; et al. Correction: Cortical organoids model early brain development disrupted by 16p11.2 copy number variants in autism. Mol. Psychiatry 2021, 1, 96. [Google Scholar] [CrossRef] [PubMed]
- Rotaru, D.C.; Mientjes, E.J.; Elgersma, Y. Angelman Syndrome: From Mouse Models to Therapy. Neuroscience 2020, 445, 172–189. [Google Scholar] [CrossRef]
- Aoki, S. Biorender. Available online: https://biorender.com/ (accessed on 16 November 2021).
- Allen Institute for Brain Science. Available online: https://atlas.brain-map.org/ (accessed on 16 November 2021).
- Ting, J.T.; Feng, G. Glutamatergic Synaptic Dysfunction and Obsessive-Compulsive Disorder. Curr. Chem. Genom. 2008, 2, 62–75. [Google Scholar] [CrossRef] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Riemersma, I.W.; Havekes, R.; Kas, M.J.H. Spatial and Temporal Gene Function Studies in Rodents: Towards Gene-Based Therapies for Autism Spectrum Disorder. Genes 2022, 13, 28. https://doi.org/10.3390/genes13010028
Riemersma IW, Havekes R, Kas MJH. Spatial and Temporal Gene Function Studies in Rodents: Towards Gene-Based Therapies for Autism Spectrum Disorder. Genes. 2022; 13(1):28. https://doi.org/10.3390/genes13010028
Chicago/Turabian StyleRiemersma, Iris W., Robbert Havekes, and Martien J. H. Kas. 2022. "Spatial and Temporal Gene Function Studies in Rodents: Towards Gene-Based Therapies for Autism Spectrum Disorder" Genes 13, no. 1: 28. https://doi.org/10.3390/genes13010028