
Wnt Pathway Extracellular Components and Their Essential Roles in Bone Homeostasis


Abstract
:1. Introduction
2. Co-Receptors
2.1. LRPs
2.2. ROR
2.3. KREMEN
2.4. LGR
3. WNT Ligands
3.1. WNT1
3.2. WNT3A
3.3. WNT4
3.4. WNT5A
3.5. WNT5B
3.6. WNT7A
3.7. WNT7B
3.8. WNT9A
3.9. WNT10A
3.10. WNT10B
3.11. WNT16
4. Inhibitors
4.1. DKK
4.2. Sclerostin and SOSTdc1
4.3. SFRPs
4.4. WIF-1
5. Future and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
Abbreviations
References
- Nusse, R.; Brown, A.; Papkoff, J.; Scambler, P.; Shackleford, G.; McMahon, A.; Moon, R.; Varmus, H. A New Nomenclature for Int-1 and Related Genes: The Wnt Gene Family. Cell 1991, 64, 231. [Google Scholar] [CrossRef]
- Nusse, R.; Varmus, H.E. Wnt Genes. Cell 1992, 69, 1073–1087. [Google Scholar] [CrossRef]
- Nusse, R. Wnt Signaling in Disease and in Development. Cell Res. 2005, 15, 28–32. [Google Scholar] [CrossRef] [Green Version]
- Van Amerongen, R. Alternative Wnt Pathways and Receptors. Cold Spring Harb. Perspect. Biol. 2012, 4, a007914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nalesso, G.; Sherwood, J.; Bertrand, J.; Pap, T.; Ramachandran, M.; De Bari, C.; Pitzalis, C.; Dell’accio, F. WNT-3A Modulates Articular Chondrocyte Phenotype by Activating Both Canonical and Noncanonical Pathways. J. Cell Biol. 2011, 193, 551–564. [Google Scholar] [CrossRef] [Green Version]
- Lu, W.; Yamamoto, V.; Ortega, B.; Baltimore, D. Mammalian Ryk is a Wnt Coreceptor Required for Stimulation of Neurite Outgrowth. Cell 2004, 119, 97–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, X.; Semenov, M.; Tamai, K.; Zeng, X. LDL Receptor-Related Proteins 5 and 6 in Wnt/β-Catenin Signaling: Arrows Point the Way. Development 2004, 131, 1663–1677. [Google Scholar] [CrossRef] [Green Version]
- Oishi, I.; Suzuki, H.; Onishi, N.; Takada, R.; Kani, S.; Ohkawara, B.; Koshida, I.; Suzuki, K.; Yamada, G.; Schwabe, G.C.; et al. The Receptor Tyrosine Kinase Ror2 Is Involved in Non-Canonical Wnt5a/JNK Signalling Pathway. Genes Cells 2003, 8, 645–654. [Google Scholar] [CrossRef]
- Dann, C.E.; Hsieh, J.C.; Rattner, A.; Sharma, D.; Nathans, J.; Leahy, D.J. Insights into Wnt Binding and Signalling from the Structures of Two Frizzled Cysteine-Rich Domains. Nature 2001, 412, 86–90. [Google Scholar] [CrossRef]
- Dijksterhuis, J.P.; Petersen, J.; Schulte, G. WNT/Frizzled Signalling: Receptor-Ligand Selectivity with Focus on FZD-G Protein Signalling and Its Physiological Relevance: IUPHAR Review 3. Br. J. Pharmacol. 2014, 171, 1195–1209. [Google Scholar] [CrossRef] [Green Version]
- Liu, F.; Kohlmeier, S.; Wang, C.-Y. Wnt Signaling and Skeletal Development. Cell Signal. 2008, 20, 999–1009. [Google Scholar] [CrossRef] [Green Version]
- Baron, R.; Kneissel, M. WNT Signaling in Bone Homeostasis and Disease: From Human Mutations to Treatments. Nat. Med. 2013, 19, 179–192. [Google Scholar] [CrossRef] [PubMed]
- Karner, C.M.; Long, F. Wnt Signaling and Cellular Metabolism in Osteoblasts. Cell. Mol. Life Sci. 2017, 74, 1649–1657. [Google Scholar] [CrossRef] [PubMed]
- Cadigan, K.M.; Peifer, M. Wnt Signaling from Development to Disease: Insights from Model Systems. Cold Spring Harb. Perspect. Biol. 2009, 1, a002881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jenny, A. Planar Cell Polarity Signaling in the Drosophila Eye. Curr. Top. Dev. Biol. 2010, 93, 189–227. [Google Scholar] [PubMed] [Green Version]
- Kohn, A.D.; Moon, R.T. Wnt and Calcium Signaling: Β-Catenin-Independent Pathways. Cell Calcium 2005, 38, 439–446. [Google Scholar] [CrossRef] [PubMed]
- Glass, D.A., II; Karsenty, G. Molecular Bases of the Regulation of Bone Remodeling by the Canonical Wnt Signaling Pathway. Curr. Top. Dev. Biol. 2006, 73, 43–84. [Google Scholar]
- Tu, X.; Joeng, K.S.; Nakayama, K.I.; Nakayama, K.; Rajagopal, J.; Carroll, T.J.; McMahon, A.P.; Long, F. Noncanonical Wnt Signaling through G Protein-Linked PKCδ Activation Promotes Bone Formation. Dev. Cell 2007, 12, 113–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, S.-Y.; Chen, C.-L.; Wu, Y.-L.; Yang, Y.-C.; Hwu, Y.-M. Ratio of Wnt3a to BMP4 Doses is Critical to Their Synergistic Effects on Proliferation of Differentiating Mouse Embryonic Stem Cells. Cell Prolif. 2008, 41, 492–505. [Google Scholar] [CrossRef]
- Qiu, W.; Chen, L.; Kassem, M. Activation of Non-Canonical Wnt/JNK Pathway by Wnt3a Is Associated with Differentiation Fate Determination of Human Bone Marrow Stromal (mesenchymal) Stem Cells. Biochem. Biophys. Res. Commun. 2011, 413, 98–104. [Google Scholar] [CrossRef]
- Qu, F.; Wang, J.; Xu, N.; Liu, C.; Li, S.; Wang, N.; Qi, W.; Li, H.; Li, C.; Geng, Z.; et al. WNT3A Modulates Chondrogenesis via Canonical and Non-Canonical Wnt Pathways in MSCs. Front. Biosci. 2013, 18, 493–503. [Google Scholar]
- Esen, E.; Chen, J.; Karner, C.M.; Okunade, A.L.; Patterson, B.W.; Long, F. WNT-LRP5 Signaling Induces Warburg Effect through mTORC2 Activation during Osteoblast Differentiation. Cell Metab. 2013, 17, 745–755. [Google Scholar] [CrossRef] [Green Version]
- Luga, V.; Zhang, L.; Viloria-Petit, A.M.; Ogunjimi, A.A.; Inanlou, M.R.; Chiu, E.; Buchanan, M.; Hosein, A.N.; Basik, M.; Wrana, J.L. Exosomes Mediate Stromal Mobilization of Autocrine Wnt-PCP Signaling in Breast Cancer Cell Migration. Cell 2012, 151, 1542–1556. [Google Scholar] [CrossRef] [Green Version]
- Mentink, R.A.; Rella, L.; Radaszkiewicz, T.W.; Gybel, T.; Betist, M.C.; Bryja, V.; Korswagen, H.C. The Planar Cell Polarity Protein VANG-1/Vangl Negatively Regulates Wnt/β-Catenin Signaling through a Dvl Dependent Mechanism. PLoS Genet. 2018, 14, e1007840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Billiard, J.; Way, D.S.; Seestaller-Wehr, L.M.; Moran, R.A.; Mangine, A.; Bodine, P.V.N. The Orphan Receptor Tyrosine Kinase Ror2 Modulates Canonical Wnt Signaling in Osteoblastic Cells. Mol. Endocrinol. 2005, 19, 90–101. [Google Scholar] [CrossRef] [Green Version]
- Duchartre, Y.; Kim, Y.-M.; Kahn, M. The Wnt Signaling Pathway in Cancer. Crit. Rev. Oncol. Hematol. 2016, 99, 141–149. [Google Scholar] [CrossRef] [Green Version]
- Kawano, Y.; Kypta, R. Secreted Antagonists of the Wnt Signalling Pathway. J. Cell Sci. 2003, 116, 2627–2634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piters, E.; Boudin, E.; Van Hul, W. Wnt Signaling: A Win for Bone. Arch. Biochem. Biophys. 2008, 473, 112–116. [Google Scholar] [CrossRef]
- Martin-Orozco, E.; Sanchez-Fernandez, A.; Ortiz-Parra, I.; Ayala-San Nicolas, M. WNT Signaling in Tumors: The Way to Evade Drugs and Immunity. Front. Immunol. 2019, 10, 2854. [Google Scholar] [CrossRef]
- Wen, X.; Wu, Y.; Awadasseid, A.; Tanaka, Y.; Zhang, W. New Advances in Canonical Wnt/β-Catenin Signaling in Cancer. Cancer Manag. Res. 2020, 12, 6987–6998. [Google Scholar] [CrossRef] [PubMed]
- Doherty, L.; Sanjay, A. LGRs in Skeletal Tissues: An Emerging Role for Wnt-Associated Adult Stem Cell Markers in Bone. JBMR Plus 2020, 4, e10380. [Google Scholar] [CrossRef] [PubMed]
- Takada, R.; Satomi, Y.; Kurata, T.; Ueno, N.; Norioka, S.; Kondoh, H.; Takao, T.; Takada, S. Monounsaturated Fatty Acid Modification of Wnt Protein: Its Role in Wnt Secretion. Dev. Cell 2006, 11, 791–801. [Google Scholar] [CrossRef] [Green Version]
- Bänziger, C.; Soldini, D.; Schütt, C.; Zipperlen, P.; Hausmann, G.; Basler, K. Wntless, a Conserved Membrane Protein Dedicated to the Secretion of Wnt Proteins from Signaling Cells. Cell 2006, 125, 509–522. [Google Scholar] [CrossRef] [Green Version]
- Zhan, T.; Rindtorff, N.; Boutros, M. Wnt Signaling in Cancer. Oncogene 2017, 36, 1461–1473. [Google Scholar] [CrossRef] [PubMed]
- Keupp, K.; Beleggia, F.; Kayserili, H.; Barnes, A.M.; Steiner, M.; Semler, O.; Fischer, B.; Yigit, G.; Janda, C.Y.; Becker, J.; et al. Mutations in WNT1 Cause Different Forms of Bone Fragility. Am. J. Hum. Genet. 2013, 92, 565–574. [Google Scholar] [CrossRef] [Green Version]
- Fahiminiya, S.; Majewski, J.; Mort, J.; Moffatt, P.; Glorieux, F.H.; Rauch, F. Mutations in WNT1 Are a Cause of Osteogenesis Imperfecta. J. Med. Genet. 2013, 50, 345–348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pyott, S.M.; Tran, T.T.; Leistritz, D.F.; Pepin, M.G.; Mendelsohn, N.J.; Temme, R.T.; Fernandez, B.A.; Elsayed, S.M.; Elsobky, E.; Verma, I.; et al. WNT1 Mutations in Families Affected by Moderately Severe and Progressive Recessive Osteogenesis Imperfecta. Am. J. Hum. Genet. 2013, 92, 590–597. [Google Scholar] [CrossRef] [Green Version]
- Gong, Y.; Slee, R.B.; Fukai, N.; Rawadi, G.; Roman-Roman, S.; Reginato, A.M.; Wang, H.; Cundy, T.; Glorieux, F.H.; Lev, D.; et al. LDL Receptor-Related Protein 5 (LRP5) Affects Bone Accrual and Eye Development. Cell 2001, 107, 513–523. [Google Scholar] [CrossRef] [Green Version]
- Ai, M.; Holmen, S.L.; Van Hul, W.; Williams, B.O.; Warman, M.L. Reduced Affinity to and Inhibition by DKK1 Form a Common Mechanism by Which High Bone Mass-Associated Missense Mutations in LRP5 Affect Canonical Wnt Signaling. Mol. Cell. Biol. 2005, 25, 4946–4955. [Google Scholar] [CrossRef] [Green Version]
- Collet, C.; Ostertag, A.; Ricquebourg, M.; Delecourt, M.; Tueur, G.; Isidor, B.; Guillot, P.; Schaefer, E.; Javier, R.-M.; Funck-Brentano, T.; et al. Primary Osteoporosis in Young Adults: Genetic Basis and Identification of Novel Variants in Causal Genes. JBMR Plus 2018, 2, 12–21. [Google Scholar] [CrossRef]
- Korvala, J.; Jüppner, H.; Mäkitie, O.; Sochett, E.; Schnabel, D.; Mora, S.; Bartels, C.F.; Warman, M.L.; Deraska, D.; Cole, W.G.; et al. Mutations in LRP5 Cause Primary Osteoporosis without Features of OI by Reducing Wnt Signaling Activity. BMC Med. Genet. 2012, 13, 26. [Google Scholar] [CrossRef] [Green Version]
- Hartikka, H.; Mäkitie, O.; Männikkö, M.; Doria, A.S.; Daneman, A.; Cole, W.G.; Ala-Kokko, L.; Sochett, E.B. Heterozygous Mutations in the LDL Receptor-Related Protein 5 (LRP5) Gene Are Associated with Primary Osteoporosis in Children. J. Bone Miner. Res. 2005, 20, 783–789. [Google Scholar] [CrossRef]
- Kim, S.J.; Bieganski, T.; Sohn, Y.B.; Kozlowski, K.; Semënov, M.; Okamoto, N.; Kim, C.H.; Ko, A.-R.; Ahn, G.H.; Choi, Y.-L.; et al. Identification of Signal Peptide Domain SOST Mutations in Autosomal Dominant Craniodiaphyseal Dysplasia. Hum. Genet. 2011, 129, 497–502. [Google Scholar] [CrossRef]
- Balemans, W.; Ebeling, M.; Patel, N.; Van Hul, E.; Olson, P.; Dioszegi, M.; Lacza, C.; Wuyts, W.; Van Den Ende, J.; Willems, P.; et al. Increased Bone Density in Sclerosteosis Is due to the Deficiency of a Novel Secreted Protein (SOST). Hum. Mol. Genet. 2001, 10, 537–543. [Google Scholar] [CrossRef] [Green Version]
- Brunkow, M.E.; Gardner, J.C.; Van Ness, J.; Paeper, B.W.; Kovacevich, B.R.; Proll, S.; Skonier, J.E.; Zhao, L.; Sabo, P.J.; Fu, Y.; et al. Bone Dysplasia Sclerosteosis Results from Loss of the SOST Gene Product, a Novel Cystine Knot-Containing Protein. Am. J. Hum. Genet. 2001, 68, 577–589. [Google Scholar] [CrossRef] [Green Version]
- Piters, E.; Culha, C.; Moester, M.; Van Bezooijen, R.; Adriaensen, D.; Mueller, T.; Weidauer, S.; Jennes, K.; de Freitas, F.; Löwik, C.; et al. First Missense Mutation in the SOST Gene Causing Sclerosteosis by Loss of Sclerostin Function. Hum. Mutat. 2010, 31, E1526–E1543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leupin, O.; Piters, E.; Halleux, C.; Hu, S.; Kramer, I.; Morvan, F.; Bouwmeester, T.; Schirle, M.; Bueno-Lozano, M.; Fuentes, F.J.R.; et al. Bone Overgrowth-Associated Mutations in the LRP4 Gene Impair Sclerostin Facilitator Function. J. Biol. Chem. 2011, 286, 19489–19500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fijalkowski, I.; Geets, E.; Steenackers, E.; Van Hoof, V.; Ramos, F.J.; Mortier, G.; Fortuna, A.M.; Van Hul, W.; Boudin, E. A Novel Domain-Specific Mutation in a Sclerosteosis Patient Suggests a Role of LRP4 as an Anchor for Sclerostin in Human Bone. J. Bone Miner. Res. 2016, 31, 874–881. [Google Scholar] [CrossRef] [Green Version]
- Balemans, W.; Patel, N.; Ebeling, M.; Van Hul, E.; Wuyts, W.; Lacza, C.; Dioszegi, M.; Dikkers, F.G.; Hildering, P.; Willems, P.J.; et al. Identification of a 52 Kb Deletion Downstream of the SOST Gene in Patients with van Buchem Disease. J. Med. Genet. 2002, 39, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Little, R.D.; Carulli, J.P.; Del Mastro, R.G.; Dupuis, J.; Osborne, M.; Folz, C.; Manning, S.P.; Swain, P.M.; Zhao, S.-C.; Eustace, B.; et al. A Mutation in the LDL Receptor-Related Protein 5 Gene Results in the Autosomal Dominant High-Bone-Mass Trait. Am. J. Hum. Genet. 2002, 70, 11–19. [Google Scholar] [CrossRef] [Green Version]
- Boyden, L.M.; Mao, J.; Belsky, J.; Mitzner, L.; Farhi, A.; Mitnick, M.A.; Wu, D.; Insogna, K.; Lifton, R.P. High Bone Density due to a Mutation in LDL-Receptor-Related Protein 5. N. Engl. J. Med. 2002, 346, 1513–1521. [Google Scholar] [CrossRef]
- Van Wesenbeeck, L.; Cleiren, E.; Gram, J.; Beals, R.K.; Bénichou, O.; Scopelliti, D.; Key, L.; Renton, T.; Bartels, C.; Gong, Y.; et al. Six Novel Missense Mutations in the LDL Receptor-Related Protein 5 (LRP5) Gene in Different Conditions with an Increased Bone Density. Am. J. Hum. Genet. 2003, 72, 763–771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whyte, M.P.; McAlister, W.H.; Zhang, F.; Bijanki, V.N.; Nenninger, A.; Gottesman, G.S.; Lin, E.L.; Huskey, M.; Duan, S.; Dahir, K.; et al. New Explanation for Autosomal Dominant High Bone Mass: Mutation of Low-Density Lipoprotein Receptor-Related Protein 6. Bone 2019, 127, 228–243. [Google Scholar] [CrossRef]
- Gregson, C.L.; Wheeler, L.; Hardcastle, S.A.; Appleton, L.H.; Addison, K.A.; Brugmans, M.; Clark, G.R.; Ward, K.A.; Paggiosi, M.; Stone, M.; et al. Mutations in Known Monogenic High Bone Mass Loci Only Explain a Small Proportion of High Bone Mass Cases. J. Bone Miner. Res. 2016, 31, 640–649. [Google Scholar] [CrossRef] [Green Version]
- Sarrión, P.; Mellibovsky, L.; Urreizti, R.; Civit, S.; Cols, N.; García-Giralt, N.; Yoskovitz, G.; Aranguren, A.; Malouf, J.; Di Gregorio, S.; et al. Genetic Analysis of High Bone Mass Cases from the BARCOS Cohort of Spanish Postmenopausal Women. PLoS ONE 2014, 9, e94607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martínez-Gil, N.; Roca-Ayats, N.; Atalay, N.; Pineda-Moncusí, M.; Garcia-Giralt, N.; Van Hul, W.; Boudin, E.; Ovejero, D.; Mellibovsky, L.; Nogués, X.; et al. Functional Assessment of Coding and Regulatory Variants From the DKK1 Locus. JBMR Plus 2020, 4, e10423. [Google Scholar] [CrossRef]
- Van Bokhoven, H.; Celli, J.; Kayserili, H.; van Beusekom, E.; Balci, S.; Brussel, W.; Skovby, F.; Kerr, B.; Percin, E.F.; Akarsu, N.; et al. Mutation of the Gene Encoding the ROR2 Tyrosine Kinase Causes Autosomal Recessive Robinow Syndrome. Nat. Genet. 2000, 25, 423–426. [Google Scholar] [CrossRef]
- Afzal, A.R.; Rajab, A.; Fenske, C.D.; Oldridge, M.; Elanko, N.; Ternes-Pereira, E.; Tüysüz, B.; Murday, V.A.; Patton, M.A.; Wilkie, A.O.; et al. Recessive Robinow Syndrome, Allelic to Dominant Brachydactyly Type B, Is Caused by Mutation of ROR2. Nat. Genet. 2000, 25, 419–422. [Google Scholar] [CrossRef] [PubMed]
- Tufan, F.; Cefle, K.; Türkmen, S.; Türkmen, A.; Zorba, U.; Dursun, M.; Oztürk, S.; Palandüz, S.; Ecder, T.; Mundlos, S.; et al. Clinical and Molecular Characterization of Two Adults with Autosomal Recessive Robinow Syndrome. Am. J. Med. Genet. A 2005, 136, 185–189. [Google Scholar] [CrossRef]
- Person, A.D.; Beiraghi, S.; Sieben, C.M.; Hermanson, S.; Neumann, A.N.; Robu, M.E.; Schleiffarth, J.R.; Billington, C.J., Jr.; van Bokhoven, H.; Hoogeboom, J.M.; et al. WNT5A Mutations in Patients with Autosomal Dominant Robinow Syndrome. Dev. Dyn. 2010, 239, 327–337. [Google Scholar]
- Kiper, P.O.S.; Simsek Kiper, P.O.; Saito, H.; Gori, F.; Unger, S.; Hesse, E.; Yamana, K.; Kiviranta, R.; Solban, N.; Liu, J.; et al. Cortical-Bone Fragility—Insights from SFRP4 Deficiency in Pyle’s Disease. N. Engl. J. Med. 2016, 374, 2553–2562. [Google Scholar] [CrossRef]
- Massink, M.P.G.; Créton, M.A.; Spanevello, F.; Fennis, W.M.M.; Cune, M.S.; Savelberg, S.M.C.; Nijman, I.J.; Maurice, M.M.; van den Boogaard, M.-J.H.; van Haaften, G. Loss-of-Function Mutations in the WNT Co-Receptor LRP6 Cause Autosomal-Dominant Oligodontia. Am. J. Hum. Genet. 2015, 97, 621–626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ockeloen, C.W.; Khandelwal, K.D.; Dreesen, K.; Ludwig, K.U.; Sullivan, R.; van Rooij, I.A.L.M.; Thonissen, M.; Swinnen, S.; Phan, M.; Conte, F.; et al. Novel Mutations in LRP6 Highlight the Role of WNT Signaling in Tooth Agenesis. Genet. Med. 2016, 18, 1158–1162. [Google Scholar] [CrossRef] [Green Version]
- Song, S.; Zhao, R.; He, H.; Zhang, J.; Feng, H.; Lin, L. WNT10A Variants Are Associated with Non-Syndromic Tooth Agenesis in the General Population. Hum. Genet. 2014, 133, 117–124. [Google Scholar] [CrossRef]
- Van den Boogaard, M.-J.; Créton, M.; Bronkhorst, Y.; van der Hout, A.; Hennekam, E.; Lindhout, D.; Cune, M.; Ploos van Amstel, H.K. Mutations in WNT10A Are Present in More than Half of Isolated Hypodontia Cases. J. Med. Genet. 2012, 49, 327–331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, P.; Yang, W.; Han, D.; Wang, X.; Guo, S.; Li, J.; Li, F.; Zhang, X.; Wong, S.-W.; Bai, B.; et al. Mutations in WNT10B Are Identified in Individuals with Oligodontia. Am. J. Hum. Genet. 2016, 99, 195–201. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Pawlik, B.; Elcioglu, N.; Aglan, M.; Kayserili, H.; Yigit, G.; Percin, F.; Goodman, F.; Nürnberg, G.; Cenani, A.; et al. LRP4 Mutations Alter Wnt/β-Catenin Signaling and Cause Limb and Kidney Malformations in Cenani-Lenz Syndrome. Am. J. Hum. Genet. 2010, 86, 696–706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sukenik Halevy, R.; Chien, H.-C.; Heinz, B.; Bamshad, M.J.; Nickerson, D.A.; University of Washington Center for Mendelian Genomics; Kircher, M.; Ahituv, N. Mutations in the Fourth β-Propeller Domain of LRP4 Are Associated with Isolated Syndactyly with Fusion of the Third and Fourth Fingers. Hum. Mutat. 2018, 39, 811–815. [Google Scholar] [CrossRef]
- Hettiaracchchi, D.; Bonnard, C.; Jayawardana, S.M.A.; Ng, A.Y.J.; Tohari, S.; Venkatesh, B.; Reversade, B.; Singaraja, R.; Dissanayake, V.H.W. Cenani-Lenz Syndactyly Syndrome—A Case Report of a Family with Isolated Syndactyly. BMC Med. Genet. 2018, 19, 125. [Google Scholar] [CrossRef] [Green Version]
- Schwabe, G.C.; Tinschert, S.; Buschow, C.; Meinecke, P.; Wolff, G.; Gillessen-Kaesbach, G.; Oldridge, M.; Wilkie, A.O.; Kömec, R.; Mundlos, S. Distinct Mutations in the Receptor Tyrosine Kinase Gene ROR2 Cause Brachydactyly Type B. Am. J. Hum. Genet. 2000, 67, 822–831. [Google Scholar] [CrossRef] [Green Version]
- Oldridge, M.; Fortuna, A.M.; Maringa, M.; Propping, P.; Mansour, S.; Pollitt, C.; DeChiara, T.M.; Kimble, R.B.; Valenzuela, D.M.; Yancopoulos, G.D.; et al. Dominant Mutations in ROR2, Encoding an Orphan Receptor Tyrosine Kinase, Cause Brachydactyly Type B. Nat. Genet. 2000, 24, 275–278. [Google Scholar] [CrossRef] [PubMed]
- Ugur, S.A.; Tolun, A. Homozygous WNT10b Mutation and Complex Inheritance in Split-Hand/Foot Malformation. Hum. Mol. Genet. 2008, 17, 2644–2653. [Google Scholar] [CrossRef]
- Intarak, N.; Theerapanon, T.; Srijunbarl, A.; Suphapeetiporn, K.; Porntaveetus, T.; Shotelersuk, V. Novel Compound Heterozygous Mutations in KREMEN1 Confirm It as a Disease Gene for Ectodermal Dysplasia. Br. J. Dermatol. 2018, 179, 758–760. [Google Scholar] [CrossRef]
- Issa, Y.A.; Kamal, L.; Rayyan, A.A.; Dweik, D.; Pierce, S.; Lee, M.K.; King, M.-C.; Walsh, T.; Kanaan, M. Mutation of KREMEN1, a Modulator of Wnt Signaling, Is Responsible for Ectodermal Dysplasia Including Oligodontia in Palestinian Families. Eur. J. Hum. Genet. 2016, 24, 1430–1435. [Google Scholar] [CrossRef] [Green Version]
- Woods, C.G.; Stricker, S.; Seemann, P.; Stern, R.; Cox, J.; Sherridan, E.; Roberts, E.; Springell, K.; Scott, S.; Karbani, G.; et al. Mutations in WNT7A Cause a Range of Limb Malformations, Including Fuhrmann Syndrome and Al-Awadi/Raas-Rothschild/Schinzel Phocomelia Syndrome. Am. J. Hum. Genet. 2006, 79, 402–408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alves, L.U.; Santos, S.; Musso, C.M.; Ezquina, S.A.; Opitz, J.M.; Kok, F.; Otto, P.A.; Mingroni-Netto, R.C. Santos Syndrome Is Caused by Mutation in the WNT7A Gene. J. Hum. Genet. 2017, 62, 1073–1078. [Google Scholar] [CrossRef] [PubMed]
- Castori, M.; Castiglia, D.; Brancati, F.; Foglio, M.; Heath, S.; Floriddia, G.; Madonna, S.; Fischer, J.; Zambruno, G. Two Families Confirm Schöpf-Schulz-Passarge Syndrome as a Discrete Entity within the WNT10A Phenotypic Spectrum. Clin. Genet. 2011, 79, 92–95. [Google Scholar] [CrossRef]
- Adaimy, L.; Chouery, E.; Megarbane, H.; Mroueh, S.; Delague, V.; Nicolas, E.; Belguith, H.; de Mazancourt, P.; Megarbane, A. Mutation in WNT10A Is Associated with an Autosomal Recessive Ectodermal Dysplasia: The Odonto-Onycho-Dermal Dysplasia. Am. J. Hum. Genet. 2007, 81, 821–828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maddirevula, S.; Alsahli, S.; Alhabeeb, L.; Patel, N.; Alzahrani, F.; Shamseldin, H.E.; Anazi, S.; Ewida, N.; Alsaif, H.S.; Mohamed, J.Y.; et al. Expanding the Phenome and Variome of Skeletal Dysplasia. Genet. Med. 2018, 20, 1609–1616. [Google Scholar] [CrossRef] [PubMed]
- Iwaniec, U.T.; Wronski, T.J.; Liu, J.; Rivera, M.F.; Arzaga, R.R.; Hansen, G.; Brommage, R. PTH Stimulates Bone Formation in Mice Deficient in Lrp5. J. Bone Miner. Res. 2007, 22, 394–402. [Google Scholar] [CrossRef] [Green Version]
- Clément-Lacroix, P.; Ai, M.; Morvan, F.; Roman-Roman, S.; Vayssière, B.; Belleville, C.; Estrera, K.; Warman, M.L.; Baron, R.; Rawadi, G. Lrp5-Independent Activation of Wnt Signaling by Lithium Chloride Increases Bone Formation and Bone Mass in Mice. Proc. Natl. Acad. Sci. USA 2005, 102, 17406–17411. [Google Scholar] [CrossRef] [Green Version]
- Yadav, V.K.; Ryu, J.-H.; Suda, N.; Tanaka, K.F.; Gingrich, J.A.; Schütz, G.; Glorieux, F.H.; Chiang, C.Y.; Zajac, J.D.; Insogna, K.L.; et al. Lrp5 Controls Bone Formation by Inhibiting Serotonin Synthesis in the Duodenum. Cell 2008, 135, 825–837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holmen, S.L.; Giambernardi, T.A.; Zylstra, C.R.; Buckner-Berghuis, B.D.; Resau, J.H.; Hess, J.F.; Glatt, V.; Bouxsein, M.L.; Ai, M.; Warman, M.L.; et al. Decreased BMD and Limb Deformities in Mice Carrying Mutations in Both Lrp5 and Lrp6. J. Bone Miner. Res. 2004, 19, 2033–2040. [Google Scholar] [CrossRef]
- Sawakami, K.; Robling, A.G.; Ai, M.; Pitner, N.D.; Liu, D.; Warden, S.J.; Li, J.; Maye, P.; Rowe, D.W.; Duncan, R.L.; et al. The Wnt Co-Receptor LRP5 Is Essential for Skeletal Mechanotransduction but Not for the Anabolic Bone Response to Parathyroid Hormone Treatment. J. Biol. Chem. 2006, 281, 23698–23711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kato, M.; Patel, M.S.; Levasseur, R.; Lobov, I.; Chang, B.H.-J.; Glass, D.A., 2nd; Hartmann, C.; Li, L.; Hwang, T.-H.; Brayton, C.F.; et al. Cbfa1-Independent Decrease in Osteoblast Proliferation, Osteopenia, and Persistent Embryonic Eye Vascularization in Mice Deficient in Lrp5, a Wnt Coreceptor. J. Cell Biol. 2002, 157, 303–314. [Google Scholar] [CrossRef] [PubMed]
- Joeng, K.S.; Schumacher, C.A.; Zylstra-Diegel, C.R.; Long, F.; Williams, B.O. Lrp5 and Lrp6 Redundantly Control Skeletal Development in the Mouse Embryo. Dev. Biol. 2011, 359, 222–229. [Google Scholar] [CrossRef] [Green Version]
- Riddle, R.C.; Diegel, C.R.; Leslie, J.M.; Van Koevering, K.K.; Faugere, M.-C.; Clemens, T.L.; Williams, B.O. Lrp5 and Lrp6 Exert Overlapping Functions in Osteoblasts during Postnatal Bone Acquisition. PLoS ONE 2013, 8, e63323. [Google Scholar] [CrossRef] [Green Version]
- Cui, Y.; Niziolek, P.J.; MacDonald, B.T.; Zylstra, C.R.; Alenina, N.; Robinson, D.R.; Zhong, Z.; Matthes, S.; Jacobsen, C.M.; Conlon, R.A.; et al. Lrp5 Functions in Bone to Regulate Bone Mass. Nat. Med. 2011, 17, 684–691. [Google Scholar] [CrossRef]
- Schumacher, C.A.; Joiner, D.M.; Less, K.D.; Drewry, M.O.; Williams, B.O. Characterization of Genetically Engineered Mouse Models Carrying Col2a1-Cre-Induced Deletions of Lrp5 And/or Lrp6. Bone Res. 2016, 4, 15042. [Google Scholar] [CrossRef] [Green Version]
- Babij, P.; Zhao, W.; Small, C.; Kharode, Y.; Yaworsky, P.J.; Bouxsein, M.L.; Reddy, P.S.; Bodine, P.V.N.; Robinson, J.A.; Bhat, B.; et al. High Bone Mass in Mice Expressing a Mutant LRP5 Gene. J. Bone Miner. Res. 2003, 18, 960–974. [Google Scholar] [CrossRef]
- Kang, K.S.; Hong, J.M.; Horan, D.J.; Lim, K.-E.; Bullock, W.A.; Bruzzaniti, A.; Hann, S.; Warman, M.L.; Robling, A.G. Induction of Lrp5 HBM-Causing Mutations in Cathepsin-K Expressing Cells Alters Bone Metabolism. Bone 2019, 120, 166–175. [Google Scholar] [CrossRef]
- Pinson, K.I.; Brennan, J.; Monkley, S.; Avery, B.J.; Skarnes, W.C. An LDL-Receptor-Related Protein Mediates Wnt Signalling in Mice. Nature 2000, 407, 535–538. [Google Scholar] [CrossRef] [PubMed]
- Kokubu, C.; Heinzmann, U.; Kokubu, T.; Sakai, N.; Kubota, T.; Kawai, M.; Wahl, M.B.; Galceran, J.; Grosschedl, R.; Ozono, K.; et al. Skeletal Defects in Ringelschwanz Mutant Mice Reveal That Lrp6 Is Required for Proper Somitogenesis and Osteogenesis. Development 2004, 131, 5469–5480. [Google Scholar] [CrossRef] [Green Version]
- Kubota, T.; Michigami, T.; Sakaguchi, N.; Kokubu, C.; Suzuki, A.; Namba, N.; Sakai, N.; Nakajima, S.; Imai, K.; Ozono, K. Lrp6 Hypomorphic Mutation Affects Bone Mass through Bone Resorption in Mice and Impairs Interaction with Mesd. J. Bone Miner. Res. 2008, 23, 1661–1671. [Google Scholar] [CrossRef] [PubMed]
- Weivoda, M.M.; Ruan, M.; Hachfeld, C.M.; Pederson, L.; Howe, A.; Davey, R.A.; Zajac, J.D.; Kobayashi, Y.; Williams, B.O.; Westendorf, J.J.; et al. Wnt Signaling Inhibits Osteoclast Differentiation by Activating Canonical and Noncanonical cAMP/PKA Pathways. J. Bone Miner. Res. 2016, 34, 1546–1548. [Google Scholar] [CrossRef] [Green Version]
- Xiong, L.; Jung, J.-U.; Wu, H.; Xia, W.-F.; Pan, J.-X.; Shen, C.; Mei, L.; Xiong, W.-C. Lrp4 in Osteoblasts Suppresses Bone Formation and Promotes Osteoclastogenesis and Bone Resorption. Proc. Natl. Acad. Sci. USA 2015, 112, 3487–3492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karner, C.M.; Dietrich, M.F.; Johnson, E.B.; Kappesser, N.; Tennert, C.; Percin, F.; Wollnik, B.; Carroll, T.J.; Herz, J. Lrp4 Regulates Initiation of Ureteric Budding and Is Crucial for Kidney Formation—A Mouse Model for Cenani-Lenz Syndrome. PLoS ONE 2010, 5, e10418. [Google Scholar] [CrossRef]
- Weatherbee, S.D.; Anderson, K.V.; Niswander, L.A. LDL-Receptor-Related Protein 4 Is Crucial for Formation of the Neuromuscular Junction. Development 2006, 133, 4993–5000. [Google Scholar] [CrossRef] [Green Version]
- Choi, H.Y.; Dieckmann, M.; Herz, J.; Niemeier, A. Lrp4, a Novel Receptor for Dickkopf 1 and Sclerostin, Is Expressed by Osteoblasts and Regulates Bone Growth and Turnover in vivo. PLoS ONE 2009, 4, e7930. [Google Scholar] [CrossRef]
- Chang, M.-K.; Kramer, I.; Huber, T.; Kinzel, B.; Guth-Gundel, S.; Leupin, O.; Kneissel, M. Disruption of Lrp4 Function by Genetic Deletion or Pharmacological Blockade Increases Bone Mass and Serum Sclerostin Levels. Proc. Natl. Acad. Sci. USA 2014, 111, E5187–E5195. [Google Scholar] [CrossRef] [Green Version]
- Bullock, W.A.; Hoggatt, A.M.; Horan, D.J.; Elmendorf, A.J.; Sato, A.Y.; Bellido, T.; Loots, G.G.; Pavalko, F.M.; Robling, A.G. Lrp4 Mediates Bone Homeostasis and Mechanotransduction through Interaction with Sclerostin In vivo. iScience 2019, 20, 205–215. [Google Scholar] [CrossRef] [Green Version]
- Boudin, E.; Yorgan, T.; Fijalkowski, I.; Sonntag, S.; Steenackers, E.; Hendrickx, G.; Peeters, S.; De Maré, A.; Vervaet, B.; Verhulst, A.; et al. The Lrp4R1170Q Homozygous Knock-In Mouse Recapitulates the Bone Phenotype of Sclerosteosis in Humans. J. Bone Miner. Res. 2017, 32, 1739–1749. [Google Scholar] [CrossRef] [Green Version]
- Maeda, K.; Kobayashi, Y.; Udagawa, N.; Uehara, S.; Ishihara, A.; Mizoguchi, T.; Kikuchi, Y.; Takada, I.; Kato, S.; Kani, S.; et al. Wnt5a-Ror2 Signaling between Osteoblast-Lineage Cells and Osteoclast Precursors Enhances Osteoclastogenesis. Nat. Med. 2012, 18, 405–412. [Google Scholar] [CrossRef]
- Takeuchi, S.; Takeda, K.; Oishi, I.; Nomi, M.; Ikeya, M.; Itoh, K.; Tamura, S.; Ueda, T.; Hatta, T.; Otani, H.; et al. Mouse Ror2 Receptor Tyrosine Kinase Is Required for the Heart Development and Limb Formation. Genes Cells 2000, 5, 71–78. [Google Scholar] [CrossRef] [PubMed]
- DeChiara, T.M.; Kimble, R.B.; Poueymirou, W.T.; Rojas, J.; Masiakowski, P.; Valenzuela, D.M.; Yancopoulos, G.D. Ror2, Encoding a Receptor-like Tyrosine Kinase, Is Required for Cartilage and Growth Plate Development. Nat. Genet. 2000, 24, 271–274. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Ng, P.Y.; Chen, R.; Hu, D.; Berry, S.; Baron, R.; Gori, F. SFRP4 Repression of the Ror2/Jnk Cascade in Osteoclasts Protects Cortical Bone from Excessive Endosteal Resorption. Proc. Natl. Acad. Sci. USA 2019, 116, 14138–14143. [Google Scholar] [CrossRef] [Green Version]
- Uehara, S.; Udagawa, N.; Mukai, H.; Ishihara, A.; Maeda, K.; Yamashita, T.; Murakami, K.; Nishita, M.; Nakamura, T.; Kato, S.; et al. Protein Kinase N3 Promotes Bone Resorption by Osteoclasts in Response to Wnt5a-Ror2 Signaling. Sci. Signal. 2017, 10, eaan0023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ellwanger, K.; Saito, H.; Clément-Lacroix, P.; Maltry, N.; Niedermeyer, J.; Lee, W.K.; Baron, R.; Rawadi, G.; Westphal, H.; Niehrs, C. Targeted Disruption of the Wnt Regulator Kremen Induces Limb Defects and High Bone Density. Mol. Cell. Biol. 2008, 28, 4875–4882. [Google Scholar] [CrossRef] [Green Version]
- Schulze, J.; Seitz, S.; Saito, H.; Schneebauer, M.; Marshall, R.P.; Baranowsky, A.; Busse, B.; Schilling, A.F.; Friedrich, F.W.; Albers, J.; et al. Negative Regulation of Bone Formation by the Transmembrane Wnt Antagonist Kremen-2. PLoS ONE 2010, 5, e10309. [Google Scholar] [CrossRef] [Green Version]
- Luo, J.; Zhou, W.; Zhou, X.; Li, D.; Weng, J.; Yi, Z.; Cho, S.G.; Li, C.; Yi, T.; Wu, X.; et al. Regulation of Bone Formation and Remodeling by G-Protein-Coupled Receptor 48. Development 2009, 136, 2747–2756. [Google Scholar] [CrossRef] [Green Version]
- Sun, P.; Jia, K.; Zheng, C.; Zhu, X.; Li, J.; He, L.; Siwko, S.; Xue, F.; Liu, M.; Luo, J. Loss of Lgr4 Inhibits Differentiation, Migration and Apoptosis, and Promotes Proliferation in Bone Mesenchymal Stem Cells. J. Cell. Physiol. 2019, 234, 10855–10867. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Yang, Z.; Ma, Y.; Yue, Z.; Lin, H.; Qu, G.; Huang, J.; Dai, W.; Li, C.; Zheng, C.; et al. LGR4 Is a Receptor for RANKL and Negatively Regulates Osteoclast Differentiation and Bone Resorption. Nat. Med. 2016, 22, 539–546. [Google Scholar] [CrossRef]
- Mazerbourg, S.; Bouley, D.M.; Sudo, S.; Klein, C.A.; Zhang, J.V.; Kawamura, K.; Goodrich, L.V.; Rayburn, H.; Tessier-Lavigne, M.; Hsueh, A.J.W. Leucine-Rich Repeat-Containing, G Protein-Coupled Receptor 4 Null Mice Exhibit Intrauterine Growth Retardation Associated with Embryonic and Perinatal Lethality. Mol. Endocrinol. 2004, 18, 2241–2254. [Google Scholar] [CrossRef] [Green Version]
- Morita, H.; Mazerbourg, S.; Bouley, D.M.; Luo, C.-W.; Kawamura, K.; Kuwabara, Y.; Baribault, H.; Tian, H.; Hsueh, A.J.W. Neonatal Lethality of LGR5 Null Mice Is Associated with Ankyloglossia and Gastrointestinal Distension. Mol. Cell. Biol. 2004, 24, 9736–9743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Snippert, H.J.; Haegebarth, A.; Kasper, M.; Jaks, V.; van Es, J.H.; Barker, N.; van de Wetering, M.; van den Born, M.; Begthel, H.; Vries, R.G.; et al. Lgr6 Marks Stem Cells in the Hair Follicle That Generate All Cell Lineages of the Skin. Science 2010, 327, 1385–1389. [Google Scholar] [CrossRef] [Green Version]
- Lehoczky, J.A.; Tabin, C.J. Lgr6 Marks Nail Stem Cells and Is Required for Digit Tip Regeneration. Proc. Natl. Acad. Sci. USA 2015, 112, 13249–13254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nykjaer, A.; Willnow, T.E. The Low-Density Lipoprotein Receptor Gene Family: A Cellular Swiss Army Knife? Trends Cell Biol. 2002, 12, 273–280. [Google Scholar] [CrossRef]
- Herz, J.; Chen, Y.; Masiulis, I.; Zhou, L. Expanding Functions of Lipoprotein Receptors. J. Lipid Res. 2009, 50, S287–S292. [Google Scholar] [CrossRef] [Green Version]
- Strickland, D.K.; Gonias, S.L.; Argraves, W.S. Diverse Roles for the LDL Receptor Family. Trends Endocrinol. Metab. 2002, 13, 66–74. [Google Scholar] [CrossRef]
- Rey, J.-P.; Ellies, D.L. Wnt Modulators in the Biotech Pipeline. Dev. Dyn. 2010, 239, 102–114. [Google Scholar] [CrossRef] [Green Version]
- Brown, S.D.; Twells, R.C.; Hey, P.J.; Cox, R.D.; Levy, E.R.; Soderman, A.R.; Metzker, M.L.; Caskey, C.T.; Todd, J.A.; Hess, J.F. Isolation and Characterization of LRP6, a Novel Member of the Low Density Lipoprotein Receptor Gene Family. Biochem. Biophys. Res. Commun. 1998, 248, 879–888. [Google Scholar] [CrossRef] [PubMed]
- Williams, B.O.; Insogna, K.L. Where Wnts Went: The Exploding Field of Lrp5 and Lrp6 Signaling in Bone. J. Bone Miner. Res. 2009, 24, 171–178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamai, K.; Zeng, X.; Liu, C.; Zhang, X.; Harada, Y.; Chang, Z.; He, X. A Mechanism for Wnt Coreceptor Activation. Mol. Cell 2004, 13, 149–156. [Google Scholar] [CrossRef]
- Chen, W.J.; Goldstein, J.L.; Brown, M.S. NPXY, a Sequence Often Found in Cytoplasmic Tails, Is Required for Coated Pit-Mediated Internalization of the Low Density Lipoprotein Receptor. J. Biol. Chem. 1990, 265, 3116–3123. [Google Scholar] [CrossRef]
- Strickland, D.K.; Ranganathan, S. Diverse Role of LDL Receptor-Related Protein in the Clearance of Proteases and in Signaling. J. Thromb. Haemost. 2003, 1, 1663–1670. [Google Scholar] [CrossRef]
- Shen, C.; Xiong, W.-C.; Mei, L. LRP4 in Neuromuscular Junction and Bone Development and Diseases. Bone 2015, 80, 101–108. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, Y.; Li, X.; Zhang, J.; Mao, J.; Li, Z.; Zheng, J.; Li, L.; Harris, S.; Wu, D. The LRP5 High-Bone-Mass G171V Mutation Disrupts LRP5 Interaction with Mesd. Mol. Cell. Biol. 2004, 24, 4677–4684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ellies, D.L.; Viviano, B.; McCarthy, J.; Rey, J.-P.; Itasaki, N.; Saunders, S.; Krumlauf, R. Bone Density Ligand, Sclerostin, Directly Interacts with LRP5 but Not LRP5G171V to Modulate Wnt Activity. J. Bone Miner. Res. 2006, 21, 1738–1749. [Google Scholar] [CrossRef] [PubMed]
- Bhat, B.M.; Allen, K.M.; Liu, W.; Graham, J.; Morales, A.; Anisowicz, A.; Lam, H.-S.; McCauley, C.; Coleburn, V.; Cain, M.; et al. Structure-Based Mutation Analysis Shows the Importance of LRP5 Β-Propeller 1 in Modulating Dkk1-Mediated Inhibition of Wnt Signaling. Gene 2007, 391, 103–112. [Google Scholar] [CrossRef]
- Semenov, M.V.; He, X. LRP5 Mutations Linked to High Bone Mass Diseases Cause Reduced LRP5 Binding and Inhibition by SOST. J. Biol. Chem. 2006, 281, 38276–38284. [Google Scholar] [CrossRef] [Green Version]
- Balemans, W.; Piters, E.; Cleiren, E.; Ai, M.; Van Wesenbeeck, L.; Warman, M.L.; Van Hul, W. The Binding between Sclerostin and LRP5 Is Altered by DKK1 and by High-Bone Mass LRP5 Mutations. Calcif. Tissue Int. 2008, 82, 445–453. [Google Scholar] [CrossRef]
- Balemans, W.; Devogelaer, J.-P.; Cleiren, E.; Piters, E.; Caussin, E.; Van Hul, W. Novel LRP5 Missense Mutation in a Patient with a High Bone Mass Phenotype Results in Decreased DKK1-Mediated Inhibition of Wnt Signaling. J. Bone Miner. Res. 2007, 22, 708–716. [Google Scholar] [CrossRef]
- Rivadeneira, F.; Styrkársdottir, U.; Estrada, K.; Halldórsson, B.V.; Hsu, Y.-H.; Richards, J.B.; Zillikens, M.C.; Kavvoura, F.K.; Amin, N.; Aulchenko, Y.S.; et al. Twenty Bone-Mineral-Density Loci Identified by Large-Scale Meta-Analysis of Genome-Wide Association Studies. Nat. Genet. 2009, 41, 1199–1206. [Google Scholar] [PubMed] [Green Version]
- Richards, J.B.; Rivadeneira, F.; Inouye, M.; Pastinen, T.M.; Soranzo, N.; Wilson, S.G.; Andrew, T.; Falchi, M.; Gwilliam, R.; Ahmadi, K.R.; et al. Bone Mineral Density, Osteoporosis, and Osteoporotic Fractures: A Genome-Wide Association Study. Lancet 2008, 371, 1505–1512. [Google Scholar] [CrossRef] [Green Version]
- Estrada, K.; Styrkarsdottir, U.; Evangelou, E.; Hsu, Y.-H.; Duncan, E.L.; Ntzani, E.E.; Oei, L.; Albagha, O.M.E.; Amin, N.; Kemp, J.P.; et al. Genome-Wide Meta-Analysis Identifies 56 Bone Mineral Density Loci and Reveals 14 Loci Associated with Risk of Fracture. Nat. Genet. 2012, 44, 491–501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kemp, J.P.; Morris, J.A.; Medina-Gomez, C.; Forgetta, V.; Warrington, N.M.; Youlten, S.E.; Zheng, J.; Gregson, C.L.; Grundberg, E.; Trajanoska, K.; et al. Identification of 153 New Loci Associated with Heel Bone Mineral Density and Functional Involvement of GPC6 in Osteoporosis. Nat. Genet. 2017, 49, 1468–1475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Medina-Gomez, C.; Kemp, J.P.; Trajanoska, K.; Luan, J.A.; Chesi, A.; Ahluwalia, T.S.; Mook-Kanamori, D.O.; Ham, A.; Hartwig, F.P.; Evans, D.S.; et al. Life-Course Genome-Wide Association Study Meta-Analysis of Total Body BMD and Assessment of Age-Specific Effects. Am. J. Hum. Genet. 2018, 102, 88–102. [Google Scholar] [CrossRef] [Green Version]
- Trajanoska, K.; Schoufour, J.D.; de Jonge, E.A.L.; Kieboom, B.C.T.; Mulder, M.; Stricker, B.H.; Voortman, T.; Uitterlinden, A.G.; Oei, E.H.G.; Ikram, M.A.; et al. Fracture Incidence and Secular Trends between 1989 and 2013 in a Population Based Cohort: The Rotterdam Study. Bone 2018, 114, 116–124. [Google Scholar] [CrossRef]
- Pei, Y.-F.; Hu, W.-Z.; Yan, M.-W.; Li, C.-W.; Liu, L.; Yang, X.-L.; Hai, R.; Wang, X.-Y.; Shen, H.; Tian, Q.; et al. Joint Study of Two Genome-Wide Association Meta-Analyses Identified 20p12.1 and 20q13.33 for Bone Mineral Density. Bone 2018, 110, 378–385. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.K. Identification of 613 New Loci Associated with Heel Bone Mineral Density and a Polygenic Risk Score for Bone Mineral Density, Osteoporosis and Fracture. PLoS ONE 2018, 13, e0200785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kichaev, G.; Bhatia, G.; Loh, P.-R.; Gazal, S.; Burch, K.; Freund, M.K.; Schoech, A.; Pasaniuc, B.; Price, A.L. Leveraging Polygenic Functional Enrichment to Improve GWAS Power. Am. J. Hum. Genet. 2019, 104, 65–75. [Google Scholar] [CrossRef] [Green Version]
- Morris, J.A.; Kemp, J.P.; Youlten, S.E.; Laurent, L.; Logan, J.G.; Chai, R.C.; Vulpescu, N.A.; Forgetta, V.; Kleinman, A.; Mohanty, S.T.; et al. An Atlas of Genetic Influences on Osteoporosis in Humans and Mice. Nat. Genet. 2019, 51, 258–266. [Google Scholar] [CrossRef]
- Zhang, L.; Choi, H.J.; Estrada, K.; Leo, P.J.; Li, J.; Pei, Y.-F.; Zhang, Y.; Lin, Y.; Shen, H.; Liu, Y.-Z.; et al. Multistage Genome-Wide Association Meta-Analyses Identified Two New Loci for Bone Mineral Density. Hum. Mol. Genet. 2014, 23, 1923–1933. [Google Scholar] [CrossRef] [Green Version]
- Akhter, M.P.; Wells, D.J.; Short, S.J.; Cullen, D.M.; Johnson, M.L.; Haynatzki, G.R.; Babij, P.; Allen, K.M.; Yaworsky, P.J.; Bex, F.; et al. Bone Biomechanical Properties in LRP5 Mutant Mice. Bone 2004, 35, 162–169. [Google Scholar] [CrossRef]
- Niziolek, P.J.; Warman, M.L.; Robling, A.G. Mechanotransduction in Bone Tissue: The A214V and G171V Mutations in Lrp5 Enhance Load-Induced Osteogenesis in a Surface-Selective Manner. Bone 2012, 51, 459–465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yadav, V.K.; Arantes, H.P.; Barros, E.R.; Lazaretti-Castro, M.; Ducy, P. Genetic Analysis of Lrp5 Function in Osteoblast Progenitors. Calcif. Tissue Int. 2010, 86, 382–388. [Google Scholar] [CrossRef]
- Kode, A.; Obri, A.; Paone, R.; Kousteni, S.; Ducy, P.; Karsenty, G. Lrp5 Regulation of Bone Mass and Serotonin Synthesis in the Gut. Nat. Med. 2014, 20, 1228–1229. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; Niziolek, P.J.; MacDonald, B.T.; Alenina, N.; Matthes, S.; Jacobsen, C.M.; Conlon, R.A.; Brommage, R.; Powell, D.R.; He, X.; et al. Reply to Lrp5 Regulation of Bone Mass and Gut Serotonin Synthesis. Nat. Med. 2014, 20, 1229–1230. [Google Scholar] [CrossRef] [PubMed]
- Lee, G.S.; Simpson, C.; Sun, B.-H.; Yao, C.; Foer, D.; Sullivan, B.; Matthes, S.; Alenina, N.; Belsky, J.; Bader, M.; et al. Measurement of Plasma, Serum, and Platelet Serotonin in Individuals with High Bone Mass and Mutations in LRP5. J. Bone Miner. Res. 2014, 29, 976–981. [Google Scholar] [CrossRef]
- Kelly, O.G.; Pinson, K.I.; Skarnes, W.C. The Wnt Co-Receptors Lrp5 and Lrp6 Are Essential for Gastrulation in Mice. Development 2004, 131, 2803–2815. [Google Scholar] [CrossRef] [Green Version]
- Wan, M.; Yang, C.; Li, J.; Wu, X.; Yuan, H.; Ma, H.; He, X.; Nie, S.; Chang, C.; Cao, X. Parathyroid Hormone Signaling through Low-Density Lipoprotein-Related Protein 6. Genes Dev. 2008, 22, 2968–2979. [Google Scholar] [CrossRef] [Green Version]
- Barik, A.; Zhang, B.; Sohal, G.S.; Xiong, W.-C.; Mei, L. Crosstalk between Agrin and Wnt Signaling Pathways in Development of Vertebrate Neuromuscular Junction. Dev. Neurobiol. 2014, 74, 828–838. [Google Scholar] [CrossRef] [PubMed]
- Johnson, E.B.; Hammer, R.E.; Herz, J. Abnormal Development of the Apical Ectodermal Ridge and Polysyndactyly in Megf7-Deficient Mice. Hum. Mol. Genet. 2005, 14, 3523–3538. [Google Scholar] [CrossRef] [PubMed]
- Ohazama, A.; Johnson, E.B.; Ota, M.S.; Choi, H.Y.; Porntaveetus, T.; Oommen, S.; Itoh, N.; Eto, K.; Gritli-Linde, A.; Herz, J.; et al. Lrp4 Modulates Extracellular Integration of Cell Signaling Pathways in Development. PLoS ONE 2008, 3, e4092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohkawara, B.; Cabrera-Serrano, M.; Nakata, T.; Milone, M.; Asai, N.; Ito, K.; Ito, M.; Masuda, A.; Ito, Y.; Engel, A.G.; et al. LRP4 Third β-Propeller Domain Mutations Cause Novel Congenital Myasthenia by Compromising Agrin-Mediated MuSK Signaling in a Position-Specific Manner. Hum. Mol. Genet. 2014, 23, 1856–1868. [Google Scholar] [CrossRef] [Green Version]
- Xiong, L.; Jung, J.-U.; Guo, H.-H.; Pan, J.-X.; Sun, X.-D.; Mei, L.; Xiong, W.-C. Osteoblastic Lrp4 Promotes Osteoclastogenesis by Regulating ATP Release and Adenosine-AR Signaling. J. Cell Biol. 2017, 216, 761–778. [Google Scholar] [CrossRef] [Green Version]
- Johnson, E.B.; Steffen, D.J.; Lynch, K.W.; Herz, J. Defective Splicing of Megf7/Lrp4, a Regulator of Distal Limb Development, in Autosomal Recessive Mulefoot Disease. Genomics 2006, 88, 600–609. [Google Scholar] [CrossRef] [Green Version]
- Drögemüller, C.; Leeb, T.; Harlizius, B.; Tammen, I.; Distl, O.; Höltershinken, M.; Gentile, A.; Duchesne, A.; Eggen, A. Congenital Syndactyly in Cattle: Four Novel Mutations in the Low Density Lipoprotein Receptor-Related Protein 4 Gene (LRP4). BMC Genet. 2007, 8, 5. [Google Scholar] [CrossRef] [Green Version]
- Simon-Chazottes, D.; Tutois, S.; Kuehn, M.; Evans, M.; Bourgade, F.; Cook, S.; Davisson, M.T.; Guénet, J.-L. Mutations in the Gene Encoding the Low-Density Lipoprotein Receptor LRP4 Cause Abnormal Limb Development in the Mouse. Genomics 2006, 87, 673–677. [Google Scholar] [CrossRef]
- Patton, M.A.; Afzal, A.R. Robinow Syndrome. J. Med. Genet. 2002, 39, 305–310. [Google Scholar] [CrossRef]
- Endo, M.; Minami, Y. Diverse Roles for the Ror-Family Receptor Tyrosine Kinases in Neurons and Glial Cells during Development and Repair of the Nervous System. Dev. Dyn. 2018, 247, 24–32. [Google Scholar] [CrossRef] [PubMed]
- Minami, Y.; Oishi, I.; Endo, M.; Nishita, M. Ror-Family Receptor Tyrosine Kinases in Noncanonical Wnt Signaling: Their Implications in Developmental Morphogenesis and Human Diseases. Dev. Dyn. 2009, 239, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Kamizaki, K.; Endo, M.; Minami, Y.; Kobayashi, Y. Role of Noncanonical Wnt Ligands and Ror-Family Receptor Tyrosine Kinases in the Development, Regeneration, and Diseases of the Musculoskeletal System. Dev. Dyn. 2020, 250, 27–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mullin, B.H.; Walsh, J.P.; Zheng, H.-F.; Brown, S.J.; Surdulescu, G.L.; Curtis, C.; Breen, G.; Dudbridge, F.; Richards, J.B.; Spector, T.D.; et al. Genome-Wide Association Study Using Family-Based Cohorts Identifies the WLS and CCDC170/ESR1 Loci as Associated with Bone Mineral Density. BMC Genom. 2016, 17, 136. [Google Scholar] [CrossRef] [Green Version]
- Nomi, M.; Oishi, I.; Kani, S.; Suzuki, H.; Matsuda, T.; Yoda, A.; Kitamura, M.; Itoh, K.; Takeuchi, S.; Takeda, K.; et al. Loss of mRor1 Enhances the Heart and Skeletal Abnormalities in mRor2-Deficient Mice: Redundant and Pleiotropic Functions of mRor1 and mRor2 Receptor Tyrosine Kinases. Mol. Cell. Biol. 2001, 21, 8329–8335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weissenböck, M.; Latham, R.; Nishita, M.; Wolff, L.I.; Ho, H.-Y.H.; Minami, Y.; Hartmann, C. Genetic Interactions between Ror2 and Wnt9a, Ror1 and Wnt9a and Ror2 and Ror1: Phenotypic Analysis of the Limb Skeleton and Palate in Compound Mutants. Genes Cells 2019, 24, 307–317. [Google Scholar] [CrossRef] [Green Version]
- Angers, S.; Moon, R.T. Proximal Events in Wnt Signal Transduction. Nat. Rev. Mol. Cell Biol. 2009, 10, 468–477. [Google Scholar] [CrossRef]
- Mao, B.; Wu, W.; Davidson, G.; Marhold, J.; Li, M.; Mechler, B.M.; Delius, H.; Hoppe, D.; Stannek, P.; Walter, C.; et al. Kremen Proteins Are Dickkopf Receptors That Regulate Wnt/β-Catenin Signalling. Nature 2002, 417, 664–667. [Google Scholar] [CrossRef]
- Mao, B.; Niehrs, C. Kremen2 Modulates Dickkopf2 Activity during Wnt/LRP6 Signaling. Gene 2003, 302, 179–183. [Google Scholar] [CrossRef]
- Zebisch, M.; Jackson, V.A.; Zhao, Y.; Jones, E.Y. Structure of the Dual-Mode Wnt Regulator Kremen1 and Insight into Ternary Complex Formation with LRP6 and Dickkopf. Structure 2016, 24, 1599–1605. [Google Scholar] [CrossRef] [Green Version]
- Mishra, S.K.; Funair, L.; Cressley, A.; Gittes, G.K.; Burns, R.C. High-Affinity Dkk1 Receptor Kremen1 Is Internalized by Clathrin-Mediated Endocytosis. PLoS ONE 2012, 7, e52190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamamoto, H.; Sakane, H.; Yamamoto, H.; Michiue, T.; Kikuchi, A. Wnt3a and Dkk1 Regulate Distinct Internalization Pathways of LRP6 to Tune the Activation of Β-Catenin Signaling. Dev. Cell 2008, 15, 37–48. [Google Scholar] [CrossRef] [PubMed]
- Hassler, C.; Cruciat, C.-M.; Huang, Y.-L.; Kuriyama, S.; Mayor, R.; Niehrs, C. Kremen Is Required for Neural Crest Induction in Xenopus and Promotes LRP6-Mediated Wnt Signaling. Development 2007, 134, 4255–4263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cselenyi, C.S.; Lee, E. Context-Dependent Activation or Inhibition of Wnt—Catenin Signaling by Kremen. Sci. Signal. 2008, 1, pe10. [Google Scholar] [CrossRef]
- Liedert, A.; Röntgen, V.; Schinke, T.; Benisch, P.; Ebert, R.; Jakob, F.; Klein-Hitpass, L.; Lennerz, J.K.; Amling, M.; Ignatius, A. Osteoblast-Specific Krm2 Overexpression and Lrp5 Deficiency Have Different Effects on Fracture Healing in Mice. PLoS ONE 2014, 9, e103250. [Google Scholar] [CrossRef] [Green Version]
- Ji, T.H.; Grossmann, M.; Ji, I. G Protein-Coupled Receptors. I. Diversity of Receptor-Ligand Interactions. J. Biol. Chem. 1998, 273, 17299–17302. [Google Scholar] [CrossRef] [Green Version]
- Garcia, M.-I. LGRs Receptors as Peculiar GPCRs Involved in Cancer. J. Stem Cell Res. Med. 2017, 2, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Hsu, S.Y.; Liang, S.-G.; Hsueh, A.J.W. Characterization of Two LGR Genes Homologous to Gonadotropin and Thyrotropin Receptors with Extracellular Leucine-Rich Repeats and a G Protein-Coupled, Seven-Transmembrane Region. Mol. Endocrinol. 1998, 12, 1830–1845. [Google Scholar] [CrossRef]
- Hsu, S.Y.; Kudo, M.; Chen, T.; Nakabayashi, K.; Bhalla, A.; van der Spek, P.J.; van Duin, M.; Hsueh, A.J. The Three Subfamilies of Leucine-Rich Repeat-Containing G Protein-Coupled Receptors (LGR): Identification of LGR6 and LGR7 and the Signaling Mechanism for LGR7. Mol. Endocrinol. 2000, 14, 1257–1271. [Google Scholar] [CrossRef]
- De Lau, W.; Barker, N.; Low, T.Y.; Koo, B.-K.; Li, V.S.W.; Teunissen, H.; Kujala, P.; Haegebarth, A.; Peters, P.J.; van de Wetering, M.; et al. Lgr5 Homologues Associate with Wnt Receptors and Mediate R-Spondin Signalling. Nature 2011, 476, 293–297. [Google Scholar] [CrossRef]
- Barker, N.; van Es, J.H.; Kuipers, J.; Kujala, P.; van den Born, M.; Cozijnsen, M.; Haegebarth, A.; Korving, J.; Begthel, H.; Peters, P.J.; et al. Identification of Stem Cells in Small Intestine and Colon by Marker Gene Lgr5. Nature 2007, 449, 1003–1007. [Google Scholar] [CrossRef] [PubMed]
- Haegebarth, A.; Clevers, H. Wnt Signaling, lgr5, and Stem Cells in the Intestine and Skin. Am. J. Pathol. 2009, 174, 715–721. [Google Scholar] [CrossRef] [Green Version]
- Barker, N.; Clevers, H. Leucine-Rich Repeat-Containing G-Protein-Coupled Receptors as Markers of Adult Stem Cells. Gastroenterology 2010, 138, 1681–1696. [Google Scholar] [CrossRef] [PubMed]
- Jaks, V.; Barker, N.; Kasper, M.; van Es, J.H.; Snippert, H.J.; Clevers, H.; Toftgård, R. Lgr5 Marks Cycling, yet Long-Lived, Hair Follicle Stem Cells. Nat. Genet. 2008, 40, 1291–1299. [Google Scholar] [CrossRef]
- Oeztuerk-Winder, F.; Guinot, A.; Ochalek, A.; Ventura, J.-J. Regulation of Human Lung Alveolar Multipotent Cells by a Novel p38α MAPK/miR-17-92 Axis. EMBO J. 2012, 31, 3431–3441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirose, K.; Shimoda, N.; Kikuchi, Y. Expression Patterns of lgr4 and lgr6 during Zebrafish Development. Gene Expr. Patterns 2011, 11, 378–383. [Google Scholar] [CrossRef]
- McDonald, T.; Wang, R.; Bailey, W.; Xie, G.; Chen, F.; Caskey, C.T.; Liu, Q. Identification and Cloning of an Orphan G Protein-Coupled Receptor of the Glycoprotein Hormone Receptor Subfamily. Biochem. Biophys. Res. Commun. 1998, 247, 266–270. [Google Scholar] [CrossRef] [PubMed]
- Weng, J.; Luo, J.; Cheng, X.; Jin, C.; Zhou, X.; Qu, J.; Tu, L.; Ai, D.; Li, D.; Wang, J.; et al. Deletion of G Protein-Coupled Receptor 48 Leads to Ocular Anterior Segment Dysgenesis (ASD) through down-Regulation of Pitx2. Proc. Natl. Acad. Sci. USA 2008, 105, 6081–6086. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Huang, Q.; Yang, R.; Guo, X.; Dai, Y.; Zeng, J.; Zeng, Y.; Tao, L.; Li, X.; Zhou, H.; et al. Targeted next Generation Sequencing of Nine Osteoporosis-Related Genes in the Wnt Signaling Pathway among Chinese Postmenopausal Women. Endocrine 2020, 68, 669–678. [Google Scholar] [CrossRef]
- Styrkarsdottir, U.; Thorleifsson, G.; Sulem, P.; Gudbjartsson, D.F.; Sigurdsson, A.; Jonasdottir, A.; Jonasdottir, A.; Oddsson, A.; Helgason, A.; Magnusson, O.T.; et al. Nonsense Mutation in the LGR4 Gene Is Associated with Several Human Diseases and Other Traits. Nature 2013, 497, 517–520. [Google Scholar] [CrossRef]
- Krönke, G.; Uderhardt, S.; Kim, K.-A.; Stock, M.; Scholtysek, C.; Zaiss, M.M.; Surmann-Schmitt, C.; Luther, J.; Katzenbeisser, J.; David, J.-P.; et al. R-Spondin 1 Protects against Inflammatory Bone Damage during Murine Arthritis by Modulating the Wnt Pathway. Arthritis Rheum. 2010, 62, 2303–2312. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.; Zheng, X.-F.; Yang, Y.-H.; Li, B.; Wang, Y.-R.; Jiang, S.-D.; Jiang, L.-S. LGR4 Acts as a Key Receptor for R-Spondin 2 to Promote Osteogenesis through Wnt Signaling Pathway. Cell Signal. 2016, 28, 989–1000. [Google Scholar] [CrossRef]
- Zhang, M.; Zhang, P.; Liu, Y.; Lv, L.; Zhang, X.; Liu, H.; Zhou, Y. RSPO3-LGR4 Regulates Osteogenic Differentiation Of Human Adipose-Derived Stem Cells Via ERK/FGF Signalling. Sci. Rep. 2017, 7, 42841. [Google Scholar] [CrossRef] [Green Version]
- Khedgikar, V.; Lehoczky, J.A. Evidence for Lgr6 as a Novel Marker of Osteoblastic Progenitors in Mice. JBMR Plus 2018, 3, e10075. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.-L.; Zhou, Y.-M.; Tang, D.-B.; Zhou, N.; Zheng, W.-W.; Tang, Z.-H.; Duan, C.-W.; Zheng, L.; Chen, J. LGR6 Promotes Osteogenesis by Activating the Wnt/β-Catenin Signaling Pathway. Biochem. Biophys. Res. Commun. 2019, 519, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.-M.; Yang, Y.-Y.; Jing, Y.-X.; Yuan, T.-J.; Sun, L.-H.; Tao, B.; Liu, J.-M.; Zhao, H.-Y. BMP9 Reduces Bone Loss in Ovariectomized Mice by Dual Regulation of Bone Remodeling. J. Bone Miner. Res. 2020, 35, 978–993. [Google Scholar] [CrossRef] [PubMed]
- Hirai, H.; Matoba, K.; Mihara, E.; Arimori, T.; Takagi, J. Crystal Structure of a Mammalian Wnt-Frizzled Complex. Nat. Struct. Mol. Biol. 2019, 26, 372–379. [Google Scholar] [CrossRef] [PubMed]
- Janda, C.Y.; Waghray, D.; Levin, A.M.; Thomas, C.; Garcia, K.C. Structural Basis of Wnt Recognition by Frizzled. Science 2012, 337, 59–64. [Google Scholar] [CrossRef] [Green Version]
- Speer, K.F.; Sommer, A.; Tajer, B.; Mullins, M.C.; Klein, P.S.; Lemmon, M.A. Non-Acylated Wnts Can Promote Signaling. Cell Rep. 2019, 26, 875–883.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doubravska, L.; Krausova, M.; Gradl, D.; Vojtechova, M.; Tumova, L.; Lukas, J.; Valenta, T.; Pospichalova, V.; Fafilek, B.; Plachy, J.; et al. Fatty Acid Modification of Wnt1 and Wnt3a at Serine Is Prerequisite for Lipidation at Cysteine and Is Essential for Wnt Signalling. Cell Signal. 2011, 23, 837–848. [Google Scholar] [CrossRef]
- Agostino, M.; Pohl, S.Ö.-G. Wnt Binding Affinity Prediction for Putative Frizzled-Type Cysteine-Rich Domains. Int. J. Mol. Sci. 2019, 20, 1468. [Google Scholar] [CrossRef] [Green Version]
- Chu, M.L.-H.; Ahn, V.E.; Choi, H.-J.; Daniels, D.L.; Nusse, R.; Weis, W.I. Structural Studies of Wnts and Identification of an LRP6 Binding Site. Structure 2013, 21, 1235–1242. [Google Scholar] [CrossRef] [Green Version]
- Bourhis, E.; Wang, W.; Tam, C.; Hwang, J.; Zhang, Y.; Spittler, D.; Huang, O.W.; Gong, Y.; Estevez, A.; Zilberleyb, I.; et al. Wnt Antagonists Bind through a Short Peptide to the First β-Propeller Domain of LRP5/6. Structure 2011, 19, 1433–1442. [Google Scholar] [CrossRef] [Green Version]
- Van Amerongen, R.; Mikels, A.; Nusse, R. Alternative Wnt Signaling Is Initiated by Distinct Receptors. Sci. Signal. 2008, 1, re9. [Google Scholar] [CrossRef] [Green Version]
- Van Amerongen, R.; Fuerer, C.; Mizutani, M.; Nusse, R. Wnt5a Can Both Activate and Repress Wnt/β-Catenin Signaling during Mouse Embryonic Development. Dev. Biol. 2012, 369, 101–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quarto, N.; Behr, B.; Longaker, M.T. Opposite Spectrum of Activity of Canonical Wnt Signaling in the Osteogenic Context of Undifferentiated and Differentiated Mesenchymal Cells: Implications for Tissue Engineering. Tissue Eng. Part A 2010, 16, 3185–3197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, F.; Tarkkonen, K.; Nieminen-Pihala, V.; Nagano, K.; Majidi, R.A.; Puolakkainen, T.; Rummukainen, P.; Lehto, J.; Roivainen, A.; Zhang, F.-P.; et al. Mesenchymal Cell-Derived Juxtacrine Wnt1 Signaling Regulates Osteoblast Activity and Osteoclast Differentiation. J. Bone Miner. Res. 2019, 34, 1129–1142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McMahon, A.P.; Bradley, A. The Wnt-1 (int-1) Proto-Oncogene Is Required for Development of a Large Region of the Mouse Brain. Cell 1990, 62, 1073–1085. [Google Scholar] [CrossRef]
- Joeng, K.S.; Lee, Y.-C.; Jiang, M.-M.; Bertin, T.K.; Chen, Y.; Abraham, A.M.; Ding, H.; Bi, X.; Ambrose, C.G.; Lee, B.H. The Swaying Mouse as a Model of Osteogenesis Imperfecta Caused by WNT1 Mutations. Hum. Mol. Genet. 2014, 23, 4035–4042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joeng, K.S.; Long, F. Wnt7b Can Replace Ihh to Induce Hypertrophic Cartilage Vascularization but Not Osteoblast Differentiation during Endochondral Bone Development. Bone Res. 2014, 2, 14004. [Google Scholar] [CrossRef] [Green Version]
- Luther, J.; Yorgan, T.A.; Rolvien, T.; Ulsamer, L.; Koehne, T.; Liao, N.; Keller, D.; Vollersen, N.; Teufel, S.; Neven, M.; et al. Wnt1 Is an Lrp5-Independent Bone-Anabolic Wnt Ligand. Sci. Transl. Med. 2018, 10, eaau7137. [Google Scholar] [CrossRef] [Green Version]
- Joeng, K.S.; Lee, Y.-C.; Lim, J.; Chen, Y.; Jiang, M.-M.; Munivez, E.; Ambrose, C.; Lee, B.H. Osteocyte-Specific WNT1 Regulates Osteoblast Function during Bone Homeostasis. J. Clin. Investig. 2017, 127, 2678–2688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshikawa, Y.; Fujimori, T.; McMahon, A.P.; Takada, S. Evidence That Absence of Wnt-3a Signaling Promotes Neuralization instead of Paraxial Mesoderm Development in the Mouse. Dev. Biol. 1997, 183, 234–242. [Google Scholar] [CrossRef] [Green Version]
- Takada, S.; Stark, K.L.; Shea, M.J.; Vassileva, G.; McMahon, J.A.; McMahon, A.P. Wnt-3a Regulates Somite and Tailbud Formation in the Mouse Embryo. Genes Dev. 1994, 8, 174–189. [Google Scholar] [CrossRef] [Green Version]
- Stark, K.; Vainio, S.; Vassileva, G.; McMahon, A.P. Epithelial Transformation of Metanephric Mesenchyme in the Developing Kidney Regulated by Wnt-4. Nature 1994, 372, 679–683. [Google Scholar] [CrossRef]
- Doolittle, M.L.; Calabrese, G.M.; Mesner, L.D.; Godfrey, D.A.; Maynard, R.D.; Ackert-Bicknell, C.L.; Farber, C.R. Genetic Analysis of Osteoblast Activity Identifies Zbtb40 as a Regulator of Osteoblast Activity and Bone Mass. PLoS Genet. 2020, 16, e1008805. [Google Scholar] [CrossRef]
- Yu, B.; Chang, J.; Liu, Y.; Li, J.; Kevork, K.; Al-Hezaimi, K.; Graves, D.T.; Park, N.-H.; Wang, C.-Y. Wnt4 Signaling Prevents Skeletal Aging and Inflammation by Inhibiting Nuclear Factor-κB. Nat. Med. 2014, 20, 1009–1017. [Google Scholar] [CrossRef] [Green Version]
- Später, D.; Hill, T.P.; O’sullivan, R.J.; Gruber, M.; Conner, D.A.; Hartmann, C. Wnt9a Signaling Is Required for Joint Integrity and Regulation of Ihh during Chondrogenesis. Development 2006, 133, 3039–3049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamaguchi, T.P.; Bradley, A.; McMahon, A.P.; Jones, S. A Wnt5a Pathway Underlies Outgrowth of Multiple Structures in the Vertebrate Embryo. Development 1999, 126, 1211–1223. [Google Scholar] [CrossRef] [PubMed]
- Topol, L.; Jiang, X.; Choi, H.; Garrett-Beal, L.; Carolan, P.J.; Yang, Y. Wnt-5a Inhibits the Canonical Wnt Pathway by Promoting GSK-3-Independent Β-Catenin Degradation. J. Cell Biol. 2003, 162, 899–908. [Google Scholar] [CrossRef]
- Okamoto, M.; Udagawa, N.; Uehara, S.; Maeda, K.; Yamashita, T.; Nakamichi, Y.; Kato, H.; Saito, N.; Minami, Y.; Takahashi, N.; et al. Noncanonical Wnt5a Enhances Wnt/β-Catenin Signaling during Osteoblastogenesis. Sci. Rep. 2014, 4, 4493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agalliu, D.; Takada, S.; Agalliu, I.; McMahon, A.P.; Jessell, T.M. Motor Neurons with Axial Muscle Projections Specified by Wnt4/5 Signaling. Neuron 2009, 61, 708–720. [Google Scholar] [CrossRef]
- Adamska, M.; MacDonald, B.T.; Sarmast, Z.H.; Oliver, E.R.; Meisler, M.H. En1 and Wnt7a Interact with Dkk1 during Limb Development in the Mouse. Dev. Biol. 2004, 272, 134–144. [Google Scholar] [CrossRef] [Green Version]
- Parr, B.A.; Cornish, V.A.; Cybulsky, M.I.; McMahon, A.P. Wnt7b Regulates Placental Development in Mice. Dev. Biol. 2001, 237, 324–332. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Tu, X.; Esen, E.; Joeng, K.S.; Lin, C.; Arbeit, J.M.; Rüegg, M.A.; Hall, M.N.; Ma, L.; Long, F. WNT7B Promotes Bone Formation in Part through mTORC1. PLoS Genet. 2014, 10, e1004145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, D.; He, G.; Song, F.; Wang, Z.; Liu, X.; Liao, L.; Ni, J.; Silva, M.J.; Long, F. Inducible Expression of Wnt7b Promotes Bone Formation in Aged Mice and Enhances Fracture Healing. Bone Res. 2020, 8, 4. [Google Scholar] [CrossRef] [Green Version]
- Tsukamoto, M.; Wang, K.-Y.; Tasaki, T.; Murata, Y.; Okada, Y.; Yamanaka, Y.; Nakamura, E.; Yamada, S.; Izumi, H.; Zhou, Q.; et al. Findings as a Starting Point to Unravel the Underlying Mechanisms of in vivo Interactions Involving Wnt10a in Bone, Fat and Muscle. Bone 2019, 120, 75–84. [Google Scholar] [CrossRef]
- Wang, K.-Y.; Yamada, S.; Izumi, H.; Tsukamoto, M.; Nakashima, T.; Tasaki, T.; Guo, X.; Uramoto, H.; Sasaguri, Y.; Kohno, K. Critical in vivo Roles of WNT10A in Wound Healing by Regulating Collagen Expression/synthesis in WNT10A-Deficient Mice. PLoS ONE 2018, 13, e0195156. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Wang, S.-K.; Choi, M.; Reid, B.M.; Hu, Y.; Lee, Y.-L.; Herzog, C.R.; Kim-Berman, H.; Lee, M.; Benke, P.J.; et al. Taurodontism, Variations in Tooth Number, and Misshapened Crowns in Wnt10a Null Mice and Human Kindreds. Mol. Genet. Genom. Med. 2015, 3, 40–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bennett, C.N.; Longo, K.A.; Wright, W.S.; Suva, L.J.; Lane, T.F.; Hankenson, K.D.; MacDougald, O.A. Regulation of Osteoblastogenesis and Bone Mass by Wnt10b. Proc. Natl. Acad. Sci. USA 2005, 102, 3324–3329. [Google Scholar] [CrossRef] [Green Version]
- Bennett, C.N.; Ouyang, H.; Ma, Y.L.; Zeng, Q.; Gerin, I.; Sousa, K.M.; Lane, T.F.; Krishnan, V.; Hankenson, K.D.; MacDougald, O.A. Wnt10b Increases Postnatal Bone Formation by Enhancing Osteoblast Differentiation. J. Bone Miner. Res. 2007, 22, 1924–1932. [Google Scholar] [CrossRef]
- Stevens, J.R.; Miranda-Carboni, G.A.; Singer, M.A.; Brugger, S.M.; Lyons, K.M.; Lane, T.F. Wnt10b Deficiency Results in Age-Dependent Loss of Bone Mass and Progressive Reduction of Mesenchymal Progenitor Cells. J. Bone Miner. Res. 2010, 25, 2138–2147. [Google Scholar] [CrossRef] [Green Version]
- Wergedal, J.E.; Kesavan, C.; Brommage, R.; Das, S.; Mohan, S. Role of WNT16 in the Regulation of Periosteal Bone Formation in Female Mice. Endocrinology 2015, 156, 1023–1032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Movérare-Skrtic, S.; Henning, P.; Liu, X.; Nagano, K.; Saito, H.; Börjesson, A.E.; Sjögren, K.; Windahl, S.H.; Farman, H.; Kindlund, B.; et al. Osteoblast-Derived WNT16 Represses Osteoclastogenesis and Prevents Cortical Bone Fragility Fractures. Nat. Med. 2014, 20, 1279–1288. [Google Scholar] [CrossRef] [Green Version]
- Medina-Gomez, C.; Kemp, J.P.; Estrada, K.; Eriksson, J.; Liu, J.; Reppe, S.; Evans, D.M.; Heppe, D.H.M.; Vandenput, L.; Herrera, L.; et al. Meta-Analysis of Genome-Wide Scans for Total Body BMD in Children and Adults Reveals Allelic Heterogeneity and Age-Specific Effects at the WNT16 Locus. PLoS Genet. 2012, 8, e1002718. [Google Scholar] [CrossRef] [Green Version]
- Zheng, H.-F.; Tobias, J.H.; Duncan, E.; Evans, D.M.; Eriksson, J.; Paternoster, L.; Yerges-Armstrong, L.M.; Lehtimäki, T.; Bergström, U.; Kähönen, M.; et al. WNT16 Influences Bone Mineral Density, Cortical Bone Thickness, Bone Strength, and Osteoporotic Fracture Risk. PLoS Genet. 2012, 8, e1002745. [Google Scholar] [CrossRef] [Green Version]
- Ohlsson, C.; Henning, P.; Nilsson, K.H.; Wu, J.; Gustafsson, K.L.; Sjögren, K.; Törnqvist, A.; Koskela, A.; Zhang, F.-P.; Lagerquist, M.K.; et al. Inducible Inactivation: WNT16 Regulates Cortical Bone Thickness in Adult Mice. J. Endocrinol. 2018, 237, 113–122. [Google Scholar] [CrossRef] [Green Version]
- Movérare-Skrtic, S.; Wu, J.; Henning, P.; Gustafsson, K.L.; Sjögren, K.; Windahl, S.H.; Koskela, A.; Tuukkanen, J.; Börjesson, A.E.; Lagerquist, M.K.; et al. The Bone-Sparing Effects of Estrogen and WNT16 Are Independent of Each Other. Proc. Natl. Acad. Sci. USA 2015, 112, 14972–14977. [Google Scholar] [CrossRef] [Green Version]
- Alam, I.; Alkhouli, M.; Gerard-O’Riley, R.L.; Wright, W.B.; Acton, D.; Gray, A.K.; Patel, B.; Reilly, A.M.; Lim, K.-E.; Robling, A.G.; et al. Osteoblast-Specific Overexpression of Human WNT16 Increases Both Cortical and Trabecular Bone Mass and Structure in Mice. Endocrinology 2016, 157, 722–736. [Google Scholar] [CrossRef] [PubMed]
- Alam, I.; Reilly, A.M.; Alkhouli, M.; Gerard-O’Riley, R.L.; Kasipathi, C.; Oakes, D.K.; Wright, W.B.; Acton, D.; McQueen, A.K.; Patel, B.; et al. Bone Mass and Strength Are Significantly Improved in Mice Overexpressing Human WNT16 in Osteocytes. Calcif. Tissue Int. 2017, 100, 361–373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Zhang, F.; Zeng, J.; Wu, Y.; Kemper, K.E.; Xue, A.; Zhang, M.; Powell, J.E.; Goddard, M.E.; Wray, N.R.; et al. Genotype-by-Environment Interactions Inferred from Genetic Effects on Phenotypic Variability in the UK Biobank. Sci. Adv. 2019, 5, eaaw3538. [Google Scholar] [CrossRef] [Green Version]
- Colaianni, G.; Cuscito, C.; Mongelli, T.; Pignataro, P.; Tamma, R.; Oranger, A.; Colucci, S.; Grano, M. Cellular mechanisms of bone regeneration: Role of Wnt-1 in bone-muscle interaction during physical activity 39. J. Biol. Regul. Homeost. Agents 2015, 29, 39–45. [Google Scholar]
- Chen, X.; Guo, J.; Yuan, Y.; Sun, Z.; Chen, B.; Tong, X.; Zhang, L.; Shen, C.; Zou, J. Cyclic Compression Stimulates Osteoblast Differentiation via Activation of the Wnt/β-Catenin Signaling Pathway. Mol. Med. Rep. 2017, 15, 2890–2896. [Google Scholar] [CrossRef] [PubMed]
- Holguin, N.; Brodt, M.D.; Silva, M.J. Activation of Wnt Signaling by Mechanical Loading Is Impaired in the Bone of Old Mice. J. Bone Miner. Res. 2016, 31, 2215–2226. [Google Scholar] [CrossRef] [PubMed]
- Boland, G.M.; Perkins, G.; Hall, D.J.; Tuan, R.S. Wnt 3a Promotes Proliferation and Suppresses Osteogenic Differentiation of Adult Human Mesenchymal Stem Cells. J. Cell. Biochem. 2004, 93, 1210–1230. [Google Scholar] [CrossRef]
- Shin, H.-R.; Islam, R.; Yoon, W.-J.; Lee, T.; Cho, Y.-D.; Bae, H.-S.; Kim, B.-S.; Woo, K.-M.; Baek, J.-H.; Ryoo, H.-M. Pin1-Mediated Modification Prolongs the Nuclear Retention of β-Catenin in Wnt3a-Induced Osteoblast Differentiation. J. Biol. Chem. 2016, 291, 5555–5565. [Google Scholar] [CrossRef] [Green Version]
- Boudin, E.; Fijalkowski, I.; Piters, E.; Van Hul, W. The Role of Extracellular Modulators of Canonical Wnt Signaling in Bone Metabolism and Diseases. Semin. Arthritis Rheum. 2013, 43, 220–240. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, T.; Kokabu, S.; Ohte, S.; Sasanuma, H.; Kanomata, K.; Yoneyama, K.; Kato, H.; Akita, M.; Oda, H.; Katagiri, T. Canonical Wnts and BMPs Cooperatively Induce Osteoblastic Differentiation through a GSK3β-Dependent and β-Catenin-Independent Mechanism. Differentiation 2010, 80, 46–52. [Google Scholar] [CrossRef]
- Almeida, M.; Han, L.; Bellido, T.; Manolagas, S.C.; Kousteni, S. Wnt Proteins Prevent Apoptosis of Both Uncommitted Osteoblast Progenitors and Differentiated Osteoblasts by Β-Catenin-Dependent and -Independent Signaling Cascades Involving Src/ERK and Phosphatidylinositol 3-kinase/AKT. J. Biol. Chem. 2005, 280, 41342–41351. [Google Scholar] [CrossRef] [Green Version]
- Jullien, N.; Maudinet, A.; Leloutre, B.; Ringe, J.; Häupl, T.; Marie, P.J. Downregulation of ErbB3 by Wnt3a Contributes to Wnt-Induced Osteoblast Differentiation in Mesenchymal Cells. J. Cell. Biochem. 2012, 113, 2047–2056. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Wang, H.; Jin, T.; Xu, Y.; Mei, L.; Yang, J. TLR4 Activation Promotes Bone Marrow MSC Proliferation and Osteogenic Differentiation via Wnt3a and Wnt5a Signaling. PLoS ONE 2016, 11, e0149876. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Vijayakumar, S.; Grumolato, L.; Arroyave, R.; Qiao, H.; Akiri, G.; Aaronson, S.A. Canonical Wnts Function as Potent Regulators of Osteogenesis by Human Mesenchymal Stem Cells. J. Cell Biol. 2009, 185, 67–75. [Google Scholar] [CrossRef]
- Wu, H.-Y.; Bi, R.; Sun, T.; Xie, F. Deletion of Dicer Blocks Osteogenic Differentiation via the Inhibition of Wnt Signalling. Mol. Med. Rep. 2019, 19, 2897–2905. [Google Scholar] [CrossRef]
- Chang, J.; Sonoyama, W.; Wang, Z.; Jin, Q.; Zhang, C.; Krebsbach, P.H.; Giannobile, W.; Shi, S.; Wang, C.-Y. Noncanonical Wnt-4 Signaling Enhances Bone Regeneration of Mesenchymal Stem Cells in Craniofacial Defects through Activation of p38 MAPK. J. Biol. Chem. 2007, 282, 30938–30948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kemp, J.P.; Medina-Gomez, C.; Estrada, K.; St Pourcain, B.; Heppe, D.H.M.; Warrington, N.M.; Oei, L.; Ring, S.M.; Kruithof, C.J.; Timpson, N.J.; et al. Phenotypic Dissection of Bone Mineral Density Reveals Skeletal Site Specificity and Facilitates the Identification of Novel Loci in the Genetic Regulation of Bone Mass Attainment. PLoS Genet. 2014, 10, e1004423. [Google Scholar] [CrossRef] [Green Version]
- Mikels, A.J.; Nusse, R. Purified Wnt5a Protein Activates or Inhibits Β-Catenin-TCF Signaling Depending on Receptor Context. PLoS Biol. 2006, 4, e115. [Google Scholar] [CrossRef] [PubMed]
- Bolzoni, M.; Donofrio, G.; Storti, P.; Guasco, D.; Toscani, D.; Lazzaretti, M.; Bonomini, S.; Agnelli, L.; Capocefalo, A.; Dalla Palma, B.; et al. Myeloma Cells Inhibit Non-Canonical Wnt Co-Receptor ror2 Expression in Human Bone Marrow Osteoprogenitor Cells: Effect of wnt5a/ror2 Pathway Activation on the Osteogenic Differentiation Impairment Induced by Myeloma Cells. Leukemia 2013, 27, 451–463. [Google Scholar] [CrossRef]
- Maeda, K.; Kobayashi, Y.; Koide, M.; Uehara, S.; Okamoto, M.; Ishihara, A.; Kayama, T.; Saito, M.; Marumo, K. The Regulation of Bone Metabolism and Disorders by Wnt Signaling. Int. J. Mol. Sci. 2019, 20, 5525. [Google Scholar] [CrossRef] [Green Version]
- Ishitani, T.; Kishida, S.; Hyodo-Miura, J.; Ueno, N.; Yasuda, J.; Waterman, M.; Shibuya, H.; Moon, R.T.; Ninomiya-Tsuji, J.; Matsumoto, K. The TAK1-NLK Mitogen-Activated Protein Kinase Cascade Functions in the Wnt-5a/Ca2 Pathway To Antagonize Wnt/β-Catenin Signaling. Mol. Cell. Biol. 2003, 23, 131–139. [Google Scholar] [CrossRef] [Green Version]
- Weske, S.; Vaidya, M.; Reese, A.; von Wnuck Lipinski, K.; Keul, P.; Bayer, J.K.; Fischer, J.W.; Flögel, U.; Nelsen, J.; Epple, M.; et al. Targeting Sphingosine-1-Phosphate Lyase as an Anabolic Therapy for Bone Loss. Nat. Med. 2018, 24, 667–678. [Google Scholar] [CrossRef]
- Bilkovski, R.; Schulte, D.M.; Oberhauser, F.; Gomolka, M.; Udelhoven, M.; Hettich, M.M.; Roth, B.; Heidenreich, A.; Gutschow, C.; Krone, W.; et al. Role of WNT-5a in the Determination of Human Mesenchymal Stem Cells into Preadipocytes. J. Biol. Chem. 2010, 285, 6170–6178. [Google Scholar] [CrossRef] [Green Version]
- Sonomoto, K.; Yamaoka, K.; Oshita, K.; Fukuyo, S.; Zhang, X.; Nakano, K.; Okada, Y.; Tanaka, Y. Interleukin-1β Induces Differentiation of Human Mesenchymal Stem Cells into Osteoblasts via the Wnt-5a/receptor Tyrosine Kinase-like Orphan Receptor 2 Pathway. Arthritis Rheum. 2012, 64, 3355–3363. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Topol, L.; Lee, H.; Wu, J. Wnt5a and Wnt5b Exhibit Distinct Activities in Coordinating Chondrocyte Proliferation and Differentiation. Development 2003, 130, 1003–1015. [Google Scholar] [CrossRef] [Green Version]
- Fazzi, R.; Pacini, S.; Carnicelli, V.; Trombi, L.; Montali, M.; Lazzarini, E.; Petrini, M. Mesodermal Progenitor Cells (MPCs) Differentiate into Mesenchymal Stromal Cells (MSCs) by Activation of Wnt5/calmodulin Signalling Pathway. PLoS ONE 2011, 6, e25600. [Google Scholar] [CrossRef] [Green Version]
- Hurson, C.J.; Butler, J.S.; Keating, D.T.; Murray, D.W.; Sadlier, D.M.; O’Byrne, J.M.; Doran, P.P. Gene Expression Analysis in Human Osteoblasts Exposed to Dexamethasone Identifies Altered Developmental Pathways as Putative Drivers of Osteoporosis. BMC Musculoskelet. Disord. 2007, 8, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hopwood, B.; Tsykin, A.; Findlay, D.M.; Fazzalari, N.L. Microarray Gene Expression Profiling of Osteoarthritic Bone Suggests Altered Bone Remodelling, WNT and Transforming Growth Factor-Β/bone Morphogenic Protein Signalling. Arthritis Res. Ther. 2007, 9, R100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Qattan, M.M. Molecular Basis of the Clinical Features of Al-Awadi-Raas-Rothschild (limb/pelvis/uterus-Hypoplasia/aplasia) Syndrome (AARRS) and Fuhrmann Syndrome. Am. J. Med. Genet. A 2013, 161A, 2274–2280. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.-L.; Shao, J.-S.; Cai, J.; Sierra, O.L.; Towler, D.A. Msx2 Exerts Bone Anabolism via Canonical Wnt Signaling. J. Biol. Chem. 2008, 283, 20505–20522. [Google Scholar] [CrossRef] [Green Version]
- Hu, H.; Hilton, M.J.; Tu, X.; Yu, K.; Ornitz, D.M.; Long, F. Sequential Roles of Hedgehog and Wnt Signaling in Osteoblast Development. Development 2005, 132, 49–60. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, Y.; Nawata, M.; Wakitani, S. Expression Profiles and Functional Analyses of Wnt-Related Genes in Human Joint Disorders. Am. J. Pathol. 2005, 167, 97–105. [Google Scholar] [CrossRef] [Green Version]
- Usami, Y.; Gunawardena, A.T.; Iwamoto, M.; Enomoto-Iwamoto, M. Wnt Signaling in Cartilage Development and Diseases: Lessons from Animal Studies. Lab. Investig. 2016, 96, 186–196. [Google Scholar] [CrossRef] [Green Version]
- Shen, L.; Zhou, S.; Glowacki, J. Effects of Age and Gender on WNT Gene Expression in Human Bone Marrow Stromal Cells. J. Cell. Biochem. 2009, 106, 337–343. [Google Scholar] [CrossRef] [Green Version]
- Zhou, H.; Mak, W.; Kalak, R.; Street, J.; Fong-Yee, C.; Zheng, Y.; Dunstan, C.R.; Seibel, M.J. Glucocorticoid-Dependent Wnt Signaling by Mature Osteoblasts Is a Key Regulator of Cranial Skeletal Development in Mice. Development 2009, 136, 427–436. [Google Scholar] [CrossRef] [Green Version]
- Ling, I.T.; Rochard, L.; Liao, E.C. Distinct Requirements of Wls, wnt9a, wnt5b and gpc4 in Regulating Chondrocyte Maturation and Timing of Endochondral Ossification. Dev. Biol. 2017, 421, 219–232. [Google Scholar] [CrossRef] [Green Version]
- Cawthorn, W.P.; Bree, A.J.; Yao, Y.; Du, B.; Hemati, N.; Martinez-Santibañez, G.; MacDougald, O.A. Wnt6, Wnt10a and Wnt10b Inhibit Adipogenesis and Stimulate Osteoblastogenesis through a β-Catenin-Dependent Mechanism. Bone 2012, 50, 477–489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.-S.; Wu, R.; Yang, X.; Kou, S.; MacDougald, O.A.; Yu, L.; Shi, H.; Xue, B. Inhibiting DNA Methylation Switches Adipogenesis to Osteoblastogenesis by Activating Wnt10a. Sci. Rep. 2016, 6, 25283. [Google Scholar] [CrossRef] [Green Version]
- Bohring, A.; Stamm, T.; Spaich, C.; Haase, C.; Spree, K.; Hehr, U.; Hoffmann, M.; Ledig, S.; Sel, S.; Wieacker, P.; et al. WNT10A Mutations Are a Frequent Cause of a Broad Spectrum of Ectodermal Dysplasias with Sex-Biased Manifestation Pattern in Heterozygotes. Am. J. Hum. Genet. 2009, 85, 97–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shungin, D.; Haworth, S.; Divaris, K.; Agler, C.S.; Kamatani, Y.; Keun Lee, M.; Grinde, K.; Hindy, G.; Alaraudanjoki, V.; Pesonen, P.; et al. Genome-Wide Analysis of Dental Caries and Periodontitis Combining Clinical and Self-Reported Data. Nat. Commun. 2019, 10, 2773. [Google Scholar] [CrossRef] [Green Version]
- Jonsson, L.; Magnusson, T.E.; Thordarson, A.; Jonsson, T.; Geller, F.; Feenstra, B.; Melbye, M.; Nohr, E.A.; Vucic, S.; Dhamo, B.; et al. Rare and Common Variants Conferring Risk of Tooth Agenesis. J. Dent. Res. 2018, 97, 515–522. [Google Scholar] [CrossRef] [Green Version]
- Xu, M.; Horrell, J.; Snitow, M.; Cui, J.; Gochnauer, H.; Syrett, C.M.; Kallish, S.; Seykora, J.T.; Liu, F.; Gaillard, D.; et al. WNT10A Mutation Causes Ectodermal Dysplasia by Impairing Progenitor Cell Proliferation and KLF4-Mediated Differentiation. Nat. Commun. 2017, 8, 15397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.; Zhang, N.; Liu, Y.; Liu, L.; Yin, G.; En, L. Effect of Human Wnt10b Transgene Overexpression on Peri-Implant Osteogenesis in Ovariectomized Rats. Hum. Gene Ther. 2018, 29, 1416–1427. [Google Scholar] [CrossRef]
- Pederson, L.; Ruan, M.; Westendorf, J.J.; Khosla, S.; Oursler, M.J. Regulation of Bone Formation by Osteoclasts Involves Wnt/BMP Signaling and the Chemokine Sphingosine-1-Phosphate. Proc. Natl. Acad. Sci. USA 2008, 105, 20764–20769. [Google Scholar] [CrossRef] [Green Version]
- Kelly, N.H.; Schimenti, J.C.; Ross, F.P.; van der Meulen, M.C.H. Transcriptional Profiling of Cortical versus Cancellous Bone from Mechanically-Loaded Murine Tibiae Reveals Differential Gene Expression. Bone 2016, 86, 22–29. [Google Scholar] [CrossRef] [Green Version]
- Robinson, J.A.; Chatterjee-Kishore, M.; Yaworsky, P.J.; Cullen, D.M.; Zhao, W.; Li, C.; Kharode, Y.; Sauter, L.; Babij, P.; Brown, E.L.; et al. Wnt/β-Catenin Signaling Is a Normal Physiological Response to Mechanical Loading in Bone. J. Biol. Chem. 2006, 281, 31720–31728. [Google Scholar] [CrossRef]
- Wend, P.; Wend, K.; Krum, S.A.; Miranda-Carboni, G.A. The Role of WNT10B in Physiology and Disease. Acta Physiol. 2012, 204, 34–51. [Google Scholar] [CrossRef] [PubMed]
- Kantaputra, P.N.; Hutsadaloi, A.; Kaewgahya, M.; Intachai, W.; German, R.; Koparal, M.; Leethanakul, C.; Tolun, A.; Ketudat Cairns, J.R. WNT10B Mutations Associated with Isolated Dental Anomalies. Clin. Genet. 2018, 93, 992–999. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Fallen, S.; Abaan, H.O.; Hayran, M.; Gonzalez, C.; Wodajo, F.; MacDonald, T.; Toretsky, J.A.; Uren, A. Wnt10b Induces Chemotaxis of Osteosarcoma and Correlates with Reduced Survival. Pediatr. Blood Cancer 2008, 51, 349–355. [Google Scholar] [CrossRef]
- Grassi, F.; Tyagi, A.M.; Calvert, J.W.; Gambari, L.; Walker, L.D.; Yu, M.; Robinson, J.; Li, J.-Y.; Lisignoli, G.; Vaccaro, C.; et al. Hydrogen Sulfide Is a Novel Regulator of Bone Formation Implicated in the Bone Loss Induced by Estrogen Deficiency. J. Bone Miner. Res. 2016, 31, 949–963. [Google Scholar] [CrossRef]
- Jiang, Z.; Von den Hoff, J.W.; Torensma, R.; Meng, L.; Bian, Z. Wnt16 Is Involved in Intramembranous Ossification and Suppresses Osteoblast Differentiation through the Wnt/β-Catenin Pathway. J. Cell. Physiol. 2014, 229, 384–392. [Google Scholar] [CrossRef] [PubMed]
- Hendrickx, G.; Boudin, E.; Verbeek, M.; Fransen, E.; Mortier, G.; Van Hul, W. WNT16 Requires Gα Subunits as Intracellular Partners for Both Its Canonical and Non-Canonical WNT Signalling Activity in Osteoblasts. Calcif. Tissue Int. 2020, 106, 294–302. [Google Scholar] [CrossRef] [PubMed]
- Tong, W.; Zeng, Y.; Chow, D.H.K.; Yeung, W.; Xu, J.; Deng, Y.; Chen, S.; Zhao, H.; Zhang, X.; Ho, K.K.; et al. Wnt16 Attenuates Osteoarthritis Progression through a PCP/JNK-mTORC1-PTHrP Cascade. Ann. Rheum. Dis. 2019, 78, 551–561. [Google Scholar] [CrossRef]
- Shen, J.; Chen, X.; Jia, H.; Meyers, C.A.; Shrestha, S.; Asatrian, G.; Ding, C.; Tsuei, R.; Zhang, X.; Peault, B.; et al. Effects of WNT3A and WNT16 on the Osteogenic and Adipogenic Differentiation of Perivascular Stem/Stromal Cells. Tissue Eng. Part A 2018, 24, 68–80. [Google Scholar] [CrossRef]
- Sebastian, A.; Hum, N.R.; Morfin, C.; Murugesh, D.K.; Loots, G.G. Global Gene Expression Analysis Identifies Mef2c as a Potential Player in Wnt16-Mediated Transcriptional Regulation. Gene 2018, 675, 312–321. [Google Scholar] [CrossRef]
- Moayyeri, A.; Hsu, Y.-H.; Karasik, D.; Estrada, K.; Xiao, S.-M.; Nielson, C.; Srikanth, P.; Giroux, S.; Wilson, S.G.; Zheng, H.-F.; et al. Genetic Determinants of Heel Bone Properties: Genome-Wide Association Meta-Analysis and Replication in the GEFOS/GENOMOS Consortium. Hum. Mol. Genet. 2014, 23, 3054–3068. [Google Scholar] [CrossRef] [Green Version]
- Chesi, A.; Mitchell, J.A.; Kalkwarf, H.J.; Bradfield, J.P.; Lappe, J.M.; McCormack, S.E.; Gilsanz, V.; Oberfield, S.E.; Hakonarson, H.; Shepherd, J.A.; et al. A Trans-Ethnic Genome-Wide Association Study Identifies Gender-Specific Loci Influencing Pediatric aBMD and BMC at the Distal Radius. Hum. Mol. Genet. 2015, 24, 5053–5059. [Google Scholar] [CrossRef]
- Koller, D.L.; Zheng, H.-F.; Karasik, D.; Yerges-Armstrong, L.; Liu, C.-T.; McGuigan, F.; Kemp, J.P.; Giroux, S.; Lai, D.; Edenberg, H.J.; et al. Meta-Analysis of Genome-Wide Studies Identifies WNT16 and ESR1 SNPs Associated with Bone Mineral Density in Premenopausal Women. J. Bone Miner. Res. 2013, 28, 547–558. [Google Scholar] [CrossRef] [Green Version]
- Zheng, H.-F.; Forgetta, V.; Hsu, Y.-H.; Estrada, K.; Rosello-Diez, A.; Leo, P.J.; Dahia, C.L.; Park-Min, K.H.; Tobias, J.H.; Kooperberg, C.; et al. Whole-Genome Sequencing Identifies EN1 as a Determinant of Bone Density and Fracture. Nature 2015, 526, 112–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mullin, B.H.; Zhao, J.H.; Brown, S.J.; Perry, J.R.B.; Luan, J.A.; Zheng, H.-F.; Langenberg, C.; Dudbridge, F.; Scott, R.; Wareham, N.J.; et al. Genome-Wide Association Study Meta-Analysis for Quantitative Ultrasound Parameters of Bone Identifies Five Novel Loci for Broadband Ultrasound Attenuation. Hum. Mol. Genet. 2017, 26, 2791–2802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dell’accio, F.; De Bari, C.; Eltawil, N.M.; Vanhummelen, P.; Pitzalis, C. Identification of the Molecular Response of Articular Cartilage to Injury, by Microarray Screening: Wnt-16 Expression and Signaling after Injury and in Osteoarthritis. Arthritis Rheum. 2008, 58, 1410–1421. [Google Scholar] [CrossRef]
- Nalesso, G.; Thomas, B.L.; Sherwood, J.C.; Yu, J.; Addimanda, O.; Eldridge, S.E.; Thorup, A.-S.; Dale, L.; Schett, G.; Zwerina, J.; et al. WNT16 Antagonises Excessive Canonical WNT Activation and Protects Cartilage in Osteoarthritis. Ann. Rheum. Dis. 2017, 76, 218–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Törnqvist, A.E.; Grahnemo, L.; Nilsson, K.H.; Funck-Brentano, T.; Ohlsson, C.; Movérare-Skrtic, S. Wnt16 Overexpression in Osteoblasts Increases the Subchondral Bone Mass but Has No Impact on Osteoarthritis in Young Adult Female Mice. Calcif. Tissue Int. 2020, 107, 31–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kobayashi, Y.; Thirukonda, G.J.; Nakamura, Y.; Koide, M.; Yamashita, T.; Uehara, S.; Kato, H.; Udagawa, N.; Takahashi, N. Wnt16 Regulates Osteoclast Differentiation in Conjunction with Wnt5a. Biochem. Biophys. Res. Commun. 2015, 463, 1278–1283. [Google Scholar] [CrossRef]
- Todd, H.; Galea, G.L.; Meakin, L.B.; Delisser, P.J.; Lanyon, L.E.; Windahl, S.H.; Price, J.S. Wnt16 Is Associated with Age-Related Bone Loss and Estrogen Withdrawal in Murine Bone. PLoS ONE 2015, 10, e0140260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohlsson, C.; Nilsson, K.H.; Henning, P.; Wu, J.; Gustafsson, K.L.; Poutanen, M.; Lerner, U.H.; Movérare-Skrtic, S. WNT16 Overexpression Partly Protects against Glucocorticoid-Induced Bone Loss. Am. J. Physiol. Endocrinol. Metab. 2018, 314, E597–E604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alam, I.; Oakes, D.K.; Reilly, A.M.; Billingsley, C.; Sβ, S.; Gerard-O’Riley, R.L.; Acton, D.; Sato, A.; Bellido, T.; Econs, M.J. Overexpression of WNT16 Does Not Prevent Cortical Bone Loss Due to Glucocorticoid Treatment in Mice. JBMR Plus 2018, 3, e10084. [Google Scholar] [CrossRef]
- Mukhopadhyay, M.; Shtrom, S.; Rodriguez-Esteban, C.; Chen, L.; Tsukui, T.; Gomer, L.; Dorward, D.W.; Glinka, A.; Grinberg, A.; Huang, S.P.; et al. Dickkopf1 Is Required for Embryonic Head Induction and Limb Morphogenesis in the Mouse. Dev. Cell 2001, 1, 423–434. [Google Scholar] [CrossRef] [Green Version]
- Morvan, F.; Boulukos, K.; Clément-Lacroix, P.; Roman, S.R.; Suc-Royer, I.; Vayssière, B.; Ammann, P.; Martin, P.; Pinho, S.; Pognonec, P.; et al. Deletion of a Single Allele of the Dkk1 Gene Leads to an Increase in Bone Formation and Bone Mass. J. Bone Miner. Res. 2006, 21, 934–945. [Google Scholar] [CrossRef]
- MacDonald, B.T.; Joiner, D.M.; Oyserman, S.M.; Sharma, P.; Goldstein, S.A.; He, X.; Hauschka, P.V. Bone Mass Is Inversely Proportional to Dkk1 Levels in Mice. Bone 2007, 41, 331–339. [Google Scholar] [CrossRef] [Green Version]
- MacDonald, B.T.; Adamska, M.; Meisler, M.H. Hypomorphic Expression of Dkk1 in the Doubleridge Mouse: Dose Dependence and Compensatory Interactions with Lrp6. Development 2004, 131, 2543–2552. [Google Scholar] [CrossRef] [Green Version]
- Colditz, J.; Thiele, S.; Baschant, U.; Niehrs, C.; Bonewald, L.F.; Hofbauer, L.C.; Rauner, M. Postnatal Skeletal Deletion of Dickkopf-1 Increases Bone Formation and Bone Volume in Male and Female Mice, Despite Increased Sclerostin Expression. J. Bone Miner. Res. 2018, 33, 1698–1707. [Google Scholar] [CrossRef] [Green Version]
- Witcher, P.C.; Miner, S.E.; Horan, D.J.; Bullock, W.A.; Lim, K.-E.; Kang, K.S.; Adaniya, A.L.; Ross, R.D.; Loots, G.G.; Robling, A.G. Sclerostin Neutralization Unleashes the Osteoanabolic Effects of Dkk1 Inhibition. JCI Insight 2018, 3, e98673. [Google Scholar] [CrossRef]
- Li, J.; Sarosi, I.; Cattley, R.C.; Pretorius, J.; Asuncion, F.; Grisanti, M.; Morony, S.; Adamu, S.; Geng, Z.; Qiu, W.; et al. Dkk1-Mediated Inhibition of Wnt Signaling in Bone Results in Osteopenia. Bone 2006, 39, 754–766. [Google Scholar] [CrossRef]
- Yao, G.-Q.; Wu, J.-J.; Troiano, N.; Insogna, K. Targeted Overexpression of Dkk1 in Osteoblasts Reduces Bone Mass but Does Not Impair the Anabolic Response to Intermittent PTH Treatment in Mice. J. Bone Miner. Res. 2011, 29, 141–148. [Google Scholar] [CrossRef] [Green Version]
- Lewis, S.L.; Khoo, P.-L.; De Young, R.A.; Steiner, K.; Wilcock, C.; Mukhopadhyay, M.; Westphal, H.; Jamieson, R.V.; Robb, L.; Tam, P.P.L. Dkk1 and Wnt3 Interact to Control Head Morphogenesis in the Mouse. Development 2008, 135, 1791–1801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morse, A.; Ko, F.C.; McDonald, M.M.; Lee, L.R.; Schindeler, A.; van der Meulen, M.C.H.; Little, D.G. Increased Anabolic Bone Response in Dkk1 KO Mice Following Tibial Compressive Loading. Bone 2020, 131, 115054. [Google Scholar] [CrossRef]
- McDonald, M.M.; Morse, A.; Schindeler, A.; Mikulec, K.; Peacock, L.; Cheng, T.; Bobyn, J.; Lee, L.; Baldock, P.A.; Croucher, P.I.; et al. Homozygous Dkk1 Knockout Mice Exhibit High Bone Mass Phenotype Due to Increased Bone Formation. Calcif. Tissue Int. 2018, 102, 105–116. [Google Scholar] [CrossRef]
- Li, X.; Liu, P.; Liu, W.; Maye, P.; Zhang, J.; Zhang, Y.; Hurley, M.; Guo, C.; Boskey, A.; Sun, L.; et al. Dkk2 Has a Role in Terminal Osteoblast Differentiation and Mineralized Matrix Formation. Nat. Genet. 2005, 37, 945–952. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Ominsky, M.S.; Niu, Q.-T.; Sun, N.; Daugherty, B.; D’Agostin, D.; Kurahara, C.; Gao, Y.; Cao, J.; Gong, J.; et al. Targeted Deletion of the Sclerostin Gene in Mice Results in Increased Bone Formation and Bone Strength. J. Bone Miner. Res. 2008, 23, 860–869. [Google Scholar] [CrossRef] [PubMed]
- Krause, C.; Korchynskyi, O.; de Rooij, K.; Weidauer, S.E.; de Gorter, D.J.J.; van Bezooijen, R.L.; Hatsell, S.; Economides, A.N.; Mueller, T.D.; Löwik, C.W.G.M.; et al. Distinct Modes of Inhibition by Sclerostin on Bone Morphogenetic Protein and Wnt Signaling Pathways. J. Biol. Chem. 2010, 285, 41614–41626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yee, C.S.; Manilay, J.O.; Chang, J.C.; Hum, N.R.; Murugesh, D.K.; Bajwa, J.; Mendez, M.E.; Economides, A.E.; Horan, D.J.; Robling, A.G.; et al. Conditional Deletion of Sost in MSC-Derived Lineages Identifies Specific Cell-Type Contributions to Bone Mass and B-Cell Development. J. Bone Miner. Res. 2018, 33, 1748–1759. [Google Scholar] [CrossRef] [Green Version]
- Rhee, Y.; Allen, M.R.; Condon, K.; Lezcano, V.; Ronda, A.C.; Galli, C.; Olivos, N.; Passeri, G.; O’Brien, C.A.; Bivi, N.; et al. PTH Receptor Signaling in Osteocytes Governs Periosteal Bone Formation and Intracortical Remodeling. J. Bone Miner. Res. 2011, 26, 1035–1046. [Google Scholar] [CrossRef] [Green Version]
- Tu, X.; Rhee, Y.; Condon, K.W.; Bivi, N.; Allen, M.R.; Dwyer, D.; Stolina, M.; Turner, C.H.; Robling, A.G.; Plotkin, L.I.; et al. Sost Downregulation and Local Wnt Signaling Are Required for the Osteogenic Response to Mechanical Loading. Bone 2012, 50, 209–217. [Google Scholar] [CrossRef] [Green Version]
- Winkler, D.G.; Sutherland, M.K.; Geoghegan, J.C.; Yu, C.; Hayes, T.; Skonier, J.E.; Shpektor, D.; Jonas, M.; Kovacevich, B.R.; Staehling-Hampton, K.; et al. Osteocyte Control of Bone Formation via Sclerostin, a Novel BMP Antagonist. EMBO J. 2003, 22, 6267–6276. [Google Scholar] [CrossRef] [Green Version]
- Loots, G.G.; Kneissel, M.; Keller, H.; Baptist, M.; Chang, J.; Collette, N.M.; Ovcharenko, D.; Plajzer-Frick, I.; Rubin, E.M. Genomic Deletion of a Long-Range Bone Enhancer Misregulates Sclerostin in Van Buchem Disease. Genome Res. 2005, 15, 928–935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ellies, D.L.; Economou, A.; Viviano, B.; Rey, J.-P.; Paine-Saunders, S.; Krumlauf, R.; Saunders, S. Wise Regulates Bone Deposition through Genetic Interactions with Lrp5. PLoS ONE 2014, 9, e96257. [Google Scholar] [CrossRef] [PubMed]
- Satoh, W.; Gotoh, T.; Tsunematsu, Y.; Aizawa, S.; Shimono, A. SFRP1 and SFRP2 Regulate Anteroposterior Axis Elongation and Somite Segmentation during Mouse Embryogenesis. Development 2006, 133, 989–999. [Google Scholar] [CrossRef] [Green Version]
- Bodine, P.V.N.; Zhao, W.; Kharode, Y.P.; Bex, F.J.; Lambert, A.-J.; Goad, M.B.; Gaur, T.; Stein, G.S.; Lian, J.B.; Komm, B.S. The Wnt Antagonist Secreted Frizzled-Related Protein-1 Is a Negative Regulator of Trabecular Bone Formation in Adult Mice. Mol. Endocrinol. 2004, 18, 1222–1237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bodine, P.V.N.; Seestaller-Wehr, L.; Kharode, Y.P.; Bex, F.J.; Komm, B.S. Bone Anabolic Effects of Parathyroid Hormone Are Blunted by Deletion of the Wnt Antagonist Secreted Frizzled-Related Protein-1. J. Cell. Physiol. 2007, 210, 352–357. [Google Scholar] [CrossRef]
- Yao, W.; Cheng, Z.; Shahnazari, M.; Dai, W.; Johnson, M.L.; Lane, N.E. Overexpression of Secreted Frizzled-Related Protein 1 Inhibits Bone Formation and Attenuates Parathyroid Hormone Bone Anabolic Effects. J. Bone Miner. Res. 2010, 25, 190–199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morello, R.; Bertin, T.K.; Schlaubitz, S.; Shaw, C.A.; Kakuru, S.; Munivez, E.; Hermanns, P.; Chen, Y.; Zabel, B.; Lee, B. Brachy-Syndactyly Caused by Loss of SFRP2 Function. J. Cell. Physiol. 2008, 217, 127–137. [Google Scholar] [CrossRef] [Green Version]
- Haraguchi, R.; Kitazawa, R.; Mori, K.; Tachibana, R.; Kiyonari, H.; Imai, Y.; Abe, T.; Kitazawa, S. SFRP4-Dependent Wnt Signal Modulation Is Critical for Bone Remodeling during Postnatal Development and Age-Related Bone Loss. Sci. Rep. 2016, 6, 25198. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, R.; Akiyama, H.; Kimura, H.; Otsuki, B.; Shimizu, M.; Tsuboyama, T.; Nakamura, T. Osteoblast-Targeted Expression of SFRP4 in Mice Results in Low Bone Mass. J. Bone Miner. Res. 2008, 23, 271–277. [Google Scholar] [CrossRef]
- Satoh, W.; Matsuyama, M.; Takemura, H.; Aizawa, S.; Shimono, A. SFRP1, SFRP2, and SFRP5 Regulate the Wnt/β-Catenin and the Planar Cell Polarity Pathways during Early Trunk Formation in Mouse. Genesis 2008, 46, 92–103. [Google Scholar] [CrossRef]
- Kansara, M.; Tsang, M.; Kodjabachian, L.; Sims, N.A.; Trivett, M.K.; Ehrich, M.; Dobrovic, A.; Slavin, J.; Choong, P.F.M.; Simmons, P.J.; et al. Wnt Inhibitory Factor 1 Is Epigenetically Silenced in Human Osteosarcoma, and Targeted Disruption Accelerates Osteosarcomagenesis in Mice. J. Clin. Investig. 2009, 119, 837–851. [Google Scholar] [CrossRef] [Green Version]
- Schaniel, C.; Sirabella, D.; Qiu, J.; Niu, X.; Lemischka, I.R.; Moore, K.A. Wnt-Inhibitory Factor 1 Dysregulation of the Bone Marrow Niche Exhausts Hematopoietic Stem Cells. Blood 2011, 118, 2420–2429. [Google Scholar] [CrossRef] [Green Version]
- Krupnik, V.E.; Sharp, J.D.; Jiang, C.; Robison, K.; Chickering, T.W.; Amaravadi, L.; Brown, D.E.; Guyot, D.; Mays, G.; Leiby, K.; et al. Functional and Structural Diversity of the Human Dickkopf Gene Family. Gene 1999, 238, 301–313. [Google Scholar] [CrossRef]
- Niehrs, C. Function and Biological Roles of the Dickkopf Family of Wnt Modulators. Oncogene 2006, 25, 7469–7481. [Google Scholar] [CrossRef] [Green Version]
- Glinka, A.; Wu, W.; Delius, H.; Monaghan, A.P.; Blumenstock, C.; Niehrs, C. Dickkopf-1 Is a Member of a New Family of Secreted Proteins and Functions in Head Induction. Nature 1998, 391, 357–362. [Google Scholar] [CrossRef] [PubMed]
- Mao, B.; Wu, W.; Li, Y.; Hoppe, D.; Stannek, P.; Glinka, A.; Niehrs, C. LDL-Receptor-Related Protein 6 Is a Receptor for Dickkopf Proteins. Nature 2001, 411, 321–325. [Google Scholar] [CrossRef]
- Bafico, A.; Liu, G.; Yaniv, A.; Gazit, A.; Aaronson, S.A. Novel Mechanism of Wnt Signalling Inhibition Mediated by Dickkopf-1 Interaction with LRP6/Arrow. Nat. Cell Biol. 2001, 3, 683–686. [Google Scholar] [CrossRef]
- Semënov, M.V.; Tamai, K.; Brott, B.K.; Kühl, M.; Sokol, S.; He, X. Head Inducer Dickkopf-1 Is a Ligand for Wnt Coreceptor LRP6. Curr. Biol. 2001, 11, 951–961. [Google Scholar] [CrossRef] [Green Version]
- Brott, B.K.; Sokol, S.Y. Regulation of Wnt/LRP Signaling by Distinct Domains of Dickkopf Proteins. Mol. Cell. Biol. 2002, 22, 6100–6110. [Google Scholar] [CrossRef] [Green Version]
- Bourhis, E.; Tam, C.; Franke, Y.; Bazan, J.F.; Ernst, J.; Hwang, J.; Costa, M.; Cochran, A.G.; Hannoush, R.N. Reconstitution of a frizzled8.Wnt3a.LRP6 Signaling Complex Reveals Multiple Wnt and Dkk1 Binding Sites on LRP6. J. Biol. Chem. 2010, 285, 9172–9179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahn, V.E.; Chu, M.L.-H.; Choi, H.-J.; Tran, D.; Abo, A.; Weis, W.I. Structural Basis of Wnt Signaling Inhibition by Dickkopf Binding to LRP5/6. Dev. Cell 2011, 21, 862–873. [Google Scholar] [CrossRef] [Green Version]
- Patel, S.; Barkell, A.M.; Gupta, D.; Strong, S.L.; Bruton, S.; Muskett, F.W.; Addis, P.W.; Renshaw, P.S.; Slocombe, P.M.; Doyle, C.; et al. Structural and Functional Analysis of Dickkopf 4 (Dkk4): New Insights into Dkk Evolution and Regulation of Wnt Signaling by Dkk and Kremen Proteins. J. Biol. Chem. 2018, 293, 12149–12166. [Google Scholar] [CrossRef] [Green Version]
- Forget, M.-A.; Turcotte, S.; Beauseigle, D.; Godin-Ethier, J.; Pelletier, S.; Martin, J.; Tanguay, S.; Lapointe, R. The Wnt Pathway Regulator DKK1 Is Preferentially Expressed in Hormone-Resistant Breast Tumours and in Some Common Cancer Types. Br. J. Cancer 2007, 96, 646–653. [Google Scholar] [CrossRef] [Green Version]
- Qian, J.; Xie, J.; Hong, S.; Yang, J.; Zhang, L.; Han, X.; Wang, M.; Zhan, F.; Shaughnessy, J.D., Jr.; Epstein, J.; et al. Dickkopf-1 (DKK1) Is a Widely Expressed and Potent Tumor-Associated Antigen in Multiple Myeloma. Blood 2007, 110, 1587–1594. [Google Scholar] [CrossRef]
- Ke, H.Z.; Richards, W.G.; Li, X.; Ominsky, M.S. Sclerostin and Dickkopf-1 as Therapeutic Targets in Bone Diseases. Endocr. Rev. 2012, 33, 747–783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Grisanti, M.; Fan, W.; Asuncion, F.J.; Tan, H.-L.; Dwyer, D.; Han, C.-Y.; Yu, L.; Lee, J.; Lee, E.; et al. Dickkopf-1 Regulates Bone Formation in Young Growing Rodents and upon Traumatic Injury. J. Bone Miner. Res. 2011, 26, 2610–2621. [Google Scholar] [CrossRef]
- Christodoulides, C.; Laudes, M.; Cawthorn, W.P.; Schinner, S.; Soos, M.; O’Rahilly, S.; Sethi, J.K.; Vidal-Puig, A. The Wnt Antagonist Dickkopf-1 and Its Receptors Are Coordinately Regulated during Early Human Adipogenesis. J. Cell Sci. 2006, 119, 2613–2620. [Google Scholar] [CrossRef] [Green Version]
- Pietilä, I.; Ellwanger, K.; Railo, A.; Jokela, T.; Barrantes, I.D.B.; Shan, J.; Niehrs, C.; Vainio, S.J. Secreted Wnt Antagonist Dickkopf-1 Controls Kidney Papilla Development Coordinated by Wnt-7b Signalling. Dev. Biol. 2011, 353, 50–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paic, F.; Igwe, J.C.; Nori, R.; Kronenberg, M.S.; Franceschetti, T.; Harrington, P.; Kuo, L.; Shin, D.-G.; Rowe, D.W.; Harris, S.E.; et al. Identification of Differentially Expressed Genes between Osteoblasts and Osteocytes. Bone 2009, 45, 682–692. [Google Scholar] [CrossRef] [Green Version]
- Taylor, S.; Ominsky, M.S.; Hu, R.; Pacheco, E.; He, Y.D.; Brown, D.L.; Aguirre, J.I.; Wronski, T.J.; Buntich, S.; Afshari, C.A.; et al. Time-Dependent Cellular and Transcriptional Changes in the Osteoblast Lineage Associated with Sclerostin Antibody Treatment in Ovariectomized Rats. Bone 2016, 84, 148–159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohnaka, K.; Tanabe, M.; Kawate, H.; Nawata, H.; Takayanagi, R. Glucocorticoid Suppresses the Canonical Wnt Signal in Cultured Human Osteoblasts. Biochem. Biophys. Res. Commun. 2005, 329, 177–181. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.-S.; Ko, J.-Y.; Lin, C.-L.; Wu, H.-L.; Ke, H.-J.; Tai, P.-J. Knocking down Dickkopf-1 Alleviates Estrogen Deficiency Induction of Bone Loss. A Histomorphological Study in Ovariectomized Rats. Bone 2007, 40, 485–492. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, S.F.; Fouda, N.; Abbas, A.A. Serum Dickkopf-1 Level in Postmenopausal Females: Correlation with Bone Mineral Density and Serum Biochemical Markers. J. Osteoporos. 2013, 2013, 460210. [Google Scholar] [CrossRef] [Green Version]
- Ko, J.-Y.; Wang, F.-S.; Wang, C.-J.; Wong, T.; Chou, W.-Y.; Tseng, S.-L. Increased Dickkopf-1 Expression Accelerates Bone Cell Apoptosis in Femoral Head Osteonecrosis. Bone 2010, 46, 584–591. [Google Scholar] [CrossRef] [PubMed]
- Pinzone, J.J.; Hall, B.M.; Thudi, N.K.; Vonau, M.; Qiang, Y.-W.; Rosol, T.J.; Shaughnessy, J.D., Jr. The Role of Dickkopf-1 in Bone Development, Homeostasis, and Disease. Blood 2009, 113, 517–525. [Google Scholar] [CrossRef] [Green Version]
- Daoussis, D.; Andonopoulos, A.P. The Emerging Role of Dickkopf-1 in Bone Biology: Is It the Main Switch Controlling Bone and Joint Remodeling? Semin. Arthritis Rheum. 2011, 41, 170–177. [Google Scholar] [CrossRef]
- Korvala, J.; Löija, M.; Mäkitie, O.; Sochett, E.; Jüppner, H.; Schnabel, D.; Mora, S.; Cole, W.G.; Ala-Kokko, L.; Männikkö, M. Rare Variations in WNT3A and DKK1 May Predispose Carriers to Primary Osteoporosis. Eur. J. Med. Genet. 2012, 55, 515–519. [Google Scholar] [CrossRef]
- Van de Putte, R.; Wijers, C.H.W.; de Blaauw, I.; Feitz, W.F.J.; Marcelis, C.L.M.; Hakobjan, M.; Sloots, C.E.J.; van Bever, Y.; Brunner, H.G.; Roeleveld, N.; et al. Sequencing of the DKK1 Gene in Patients with Anorectal Malformations and Hypospadias. Eur. J. Pediatr. 2015, 174, 583–587. [Google Scholar] [CrossRef]
- Ntini, E.; Louloupi, A.; Liz, J.; Muino, J.M.; Marsico, A.; Ørom, U.A.V. Long ncRNA A-ROD Activates Its Target Gene DKK1 at Its Release from Chromatin. Nat. Commun. 2018, 9, 1636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robling, A.G.; Niziolek, P.J.; Baldridge, L.A.; Condon, K.W.; Allen, M.R.; Alam, I.; Mantila, S.M.; Gluhak-Heinrich, J.; Bellido, T.M.; Harris, S.E.; et al. Mechanical Stimulation of Bone in vivo Reduces Osteocyte Expression of Sost/sclerostin. J. Biol. Chem. 2008, 283, 5866–5875. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Mao, J.; Sun, L.; Liu, W.; Wu, D. Second Cysteine-Rich Domain of Dickkopf-2 Activates Canonical Wnt Signaling Pathway via LRP-6 Independently of Dishevelled. J. Biol. Chem. 2002, 277, 5977–5981. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Wang, K.; Shao, Y.; Huang, J.; Li, X.; Shan, J.; Wu, D.; Zheng, J.J. Structural Insight into the Mechanisms of Wnt Signaling Antagonism by Dkk. J. Biol. Chem. 2008, 283, 23364–23370. [Google Scholar] [CrossRef] [Green Version]
- Veverka, V.; Henry, A.J.; Slocombe, P.M.; Ventom, A.; Mulloy, B.; Muskett, F.W.; Muzylak, M.; Greenslade, K.; Moore, A.; Zhang, L.; et al. Characterization of the Structural Features and Interactions of Sclerostin: Molecular Insight into a Key Regulator of Wnt-Mediated Bone Formation. J. Biol. Chem. 2009, 284, 10890–10900. [Google Scholar] [CrossRef] [Green Version]
- Kattamuri, C.; Luedeke, D.M.; Nolan, K.; Rankin, S.A.; Greis, K.D.; Zorn, A.M.; Thompson, T.B. Members of the DAN Family Are BMP Antagonists That Form Highly Stable Noncovalent Dimers. J. Mol. Biol. 2012, 424, 313–327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Itasaki, N.; Jones, C.M.; Mercurio, S.; Rowe, A.; Domingos, P.M.; Smith, J.C.; Krumlauf, R. Wise, a Context-Dependent Activator and Inhibitor of Wnt Signalling. Development 2003, 130, 4295–4305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Zhang, Y.; Kang, H.; Liu, W.; Liu, P.; Zhang, J.; Harris, S.E.; Wu, D. Sclerostin Binds to LRP5/6 and Antagonizes Canonical Wnt Signaling. J. Biol. Chem. 2005, 280, 19883–19887. [Google Scholar] [CrossRef] [Green Version]
- Semënov, M.; Tamai, K.; He, X. SOST Is a Ligand for LRP5/LRP6 and a Wnt Signaling Inhibitor. J. Biol. Chem. 2005, 280, 26770–26775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cruciat, C.-M.; Niehrs, C. Secreted and Transmembrane Wnt Inhibitors and Activators. Cold Spring Harb. Perspect. Biol. 2013, 5, a015081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lintern, K.B.; Guidato, S.; Rowe, A.; Saldanha, J.W.; Itasaki, N. Characterization of Wise Protein and Its Molecular Mechanism to Interact with Both Wnt and BMP Signals. J. Biol. Chem. 2009, 284, 23159–23168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Bezooijen, R.L.; Svensson, J.P.; Eefting, D.; Visser, A.; van der Horst, G.; Karperien, M.; Quax, P.H.A.; Vrieling, H.; Papapoulos, S.E.; ten Dijke, P.; et al. Wnt but Not BMP Signaling Is Involved in the Inhibitory Action of Sclerostin on BMP-Stimulated Bone Formation. J. Bone Miner. Res. 2007, 22, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Van Bezooijen, R.L.; Papapoulos, S.E.; Löwik, C.W.G.M. Bone Morphogenetic Proteins and Their Antagonists: The Sclerostin Paradigm. J. Endocrinol. Investig. 2005, 28, 15–17. [Google Scholar]
- Poole, K.E.S.; van Bezooijen, R.L.; Loveridge, N.; Hamersma, H.; Papapoulos, S.E.; Löwik, C.W.; Reeve, J. Sclerostin Is a Delayed Secreted Product of Osteocytes That Inhibits Bone Formation. FASEB J. 2005, 19, 1842–1844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dallas, S.L.; Bonewald, L.F. Dynamics of the Transition from Osteoblast to Osteocyte. Ann. N. Y. Acad. Sci. 2010, 1192, 437–443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delgado-Calle, J.; Sato, A.Y.; Bellido, T. Role and Mechanism of Action of Sclerostin in Bone. Bone 2017, 96, 29–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sebastian, A.; Loots, G.G. Transcriptional Control of Sost in Bone. Bone 2017, 96, 76–84. [Google Scholar] [CrossRef] [Green Version]
- Collette, N.M.; Yee, C.S.; Murugesh, D.; Sebastian, A.; Taher, L.; Gale, N.W.; Economides, A.N.; Harland, R.M.; Loots, G.G. Sost and Its Paralog Sostdc1 Coordinate Digit Number in a Gli3-Dependent Manner. Dev. Biol. 2013, 383, 90–105. [Google Scholar] [CrossRef] [Green Version]
- Balemans, W.; Van Den Ende, J.; Freire Paes-Alves, A.; Dikkers, F.G.; Willems, P.J.; Vanhoenacker, F.; de Almeida-Melo, N.; Alves, C.F.; Stratakis, C.A.; Hill, S.C.; et al. Localization of the Gene for Sclerosteosis to the van Buchem Disease-Gene Region on Chromosome 17q12-q21. Am. J. Hum. Genet. 1999, 64, 1661–1669. [Google Scholar] [CrossRef] [Green Version]
- Van Lierop, A.H.; Hamdy, N.A.T.; van Egmond, M.E.; Bakker, E.; Dikkers, F.G.; Papapoulos, S.E. Van Buchem Disease: Clinical, Biochemical, and Densitometric Features of Patients and Disease Carriers. J. Bone Miner. Res. 2013, 28, 848–854. [Google Scholar] [CrossRef] [PubMed]
- Van Lierop, A.H.; Hamdy, N.A.; Hamersma, H.; van Bezooijen, R.L.; Power, J.; Loveridge, N.; Papapoulos, S.E. Patients with Sclerosteosis and Disease Carriers: Human Models of the Effect of Sclerostin on Bone Turnover. J. Bone Miner. Res. 2011, 26, 2804–2811. [Google Scholar] [CrossRef]
- Martínez-Gil, N.; Roca-Ayats, N.; Cozar, M.; Garcia-Giralt, N.; Ovejero, D.; Nogués, X.; Grinberg, D.; Balcells, S. Genetics and Genomics of SOST: Functional Analysis of Variants and Genomic Regulation in Osteoblasts. Int. J. Mol. Sci. 2021, 22, 489. [Google Scholar] [CrossRef] [PubMed]
- Styrkarsdottir, U.; Thorleifsson, G.; Gudjonsson, S.A.; Sigurdsson, A.; Center, J.R.; Lee, S.H.; Nguyen, T.V.; Kwok, T.C.Y.; Lee, J.S.W.; Ho, S.C.; et al. Sequence Variants in the PTCH1 Gene Associate with Spine Bone Mineral Density and Osteoporotic Fractures. Nat. Commun. 2016, 7, 10129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weivoda, M.M.; Youssef, S.J.; Oursler, M.J. Sclerostin Expression and Functions beyond the Osteocyte. Bone 2017, 96, 45–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ardawi, M.-S.M.; Al-Kadi, H.A.; Rouzi, A.A.; Qari, M.H. Determinants of Serum Sclerostin in Healthy Pre- and Postmenopausal Women. J. Bone Miner. Res. 2011, 26, 2812–2822. [Google Scholar] [CrossRef]
- Mirza, F.S.; Padhi, I.D.; Raisz, L.G.; Lorenzo, J.A. Serum Sclerostin Levels Negatively Correlate with Parathyroid Hormone Levels and Free Estrogen Index in Postmenopausal Women. J. Clin. Endocrinol. Metab. 2010, 95, 1991–1997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roforth, M.M.; Fujita, K.; McGregor, U.I.; Kirmani, S.; McCready, L.K.; Peterson, J.M.; Drake, M.T.; Monroe, D.G.; Khosla, S. Effects of Age on Bone mRNA Levels of Sclerostin and Other Genes Relevant to Bone Metabolism in Humans. Bone 2014, 59, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Mödder, U.I.; Hoey, K.A.; Amin, S.; McCready, L.K.; Achenbach, S.J.; Riggs, B.L.; Melton, L.J., 3rd; Khosla, S. Relation of Age, Gender, and Bone Mass to Circulating Sclerostin Levels in Women and Men. J. Bone Miner. Res. 2011, 26, 373–379. [Google Scholar] [CrossRef] [Green Version]
- Amrein, K.; Amrein, S.; Drexler, C.; Dimai, H.P.; Dobnig, H.; Pfeifer, K.; Tomaschitz, A.; Pieber, T.R.; Fahrleitner-Pammer, A. Sclerostin and Its Association with Physical Activity, Age, Gender, Body Composition, and Bone Mineral Content in Healthy Adults. J. Clin. Endocrinol. Metab. 2012, 97, 148–154. [Google Scholar] [CrossRef] [Green Version]
- Ota, K.; Quint, P.; Ruan, M.; Pederson, L.; Westendorf, J.J.; Khosla, S.; Oursler, M.J. Sclerostin Is Expressed in Osteoclasts from Aged Mice and Reduces Osteoclast-Mediated Stimulation of Mineralization. J. Cell. Biochem. 2013, 114, 1901–1907. [Google Scholar] [CrossRef] [Green Version]
- Hay, E.; Bouaziz, W.; Funck-Brentano, T.; Cohen-Solal, M. Sclerostin and Bone Aging: A Mini-Review. Gerontology 2016, 62, 618–623. [Google Scholar] [CrossRef]
- Sato, A.Y.; Cregor, M.; Delgado-Calle, J.; Condon, K.W.; Allen, M.R.; Peacock, M.; Plotkin, L.I.; Bellido, T. Protection From Glucocorticoid-Induced Osteoporosis by Anti-Catabolic Signaling in the Absence of Sost/Sclerostin. J. Bone Miner. Res. 2016, 31, 1791–1802. [Google Scholar] [CrossRef] [Green Version]
- Terpos, E.; Morgan, G.; Dimopoulos, M.A.; Drake, M.T.; Lentzsch, S.; Raje, N.; Sezer, O.; García-Sanz, R.; Shimizu, K.; Turesson, I.; et al. International Myeloma Working Group Recommendations for the Treatment of Multiple Myeloma-Related Bone Disease. J. Clin. Oncol. 2013, 31, 2347–2357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- García-Fontana, B.; Morales-Santana, S.; Varsavsky, M.; García-Martín, A.; García-Salcedo, J.A.; Reyes-García, R.; Muñoz-Torres, M. Sclerostin Serum Levels in Prostate Cancer Patients and Their Relationship with Sex Steroids. Osteoporos. Int. 2014, 25, 645–651. [Google Scholar] [CrossRef] [PubMed]
- Mendoza-Villanueva, D.; Zeef, L.; Shore, P. Metastatic Breast Cancer Cells Inhibit Osteoblast Differentiation through the Runx2/CBFβ-Dependent Expression of the Wnt Antagonist, Sclerostin. Breast Cancer Res. 2011, 13, R106. [Google Scholar] [CrossRef] [Green Version]
- Zhu, D.; Mackenzie, N.C.W.; Millán, J.L.; Farquharson, C.; MacRae, V.E. The Appearance and Modulation of Osteocyte Marker Expression during Calcification of Vascular Smooth Muscle Cells. PLoS ONE 2011, 6, e19595. [Google Scholar] [CrossRef] [Green Version]
- Brandenburg, V.M.; Kramann, R.; Koos, R.; Krüger, T.; Schurgers, L.; Mühlenbruch, G.; Hübner, S.; Gladziwa, U.; Drechsler, C.; Ketteler, M. Relationship between Sclerostin and Cardiovascular Calcification in Hemodialysis Patients: A Cross-Sectional Study. BMC Nephrol. 2013, 14, 219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sebastian, A.; Loots, G.G. Genetics of Sost/SOST in Sclerosteosis and van Buchem Disease Animal Models. Metabolism 2018, 80, 38–47. [Google Scholar] [CrossRef] [Green Version]
- Collette, N.M.; Genetos, D.C.; Economides, A.N.; Xie, L.; Shahnazari, M.; Yao, W.; Lane, N.E.; Harland, R.M.; Loots, G.G. Targeted Deletion of Sost Distal Enhancer Increases Bone Formation and Bone Mass. Proc. Natl. Acad. Sci. USA 2012, 109, 14092–14097. [Google Scholar] [CrossRef] [Green Version]
- Pérez-Campo, F.M.; Santurtún, A.; García-Ibarbia, C.; Pascual, M.A.; Valero, C.; Garcés, C.; Sañudo, C.; Zarrabeitia, M.T.; Riancho, J.A. Osterix and RUNX2 Are Transcriptional Regulators of Sclerostin in Human Bone. Calcif. Tissue Int. 2016, 99, 302–309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, F.; Tang, W.; So, S.; de Crombrugghe, B.; Zhang, C. Sclerostin Is a Direct Target of Osteoblast-Specific Transcription Factor Osterix. Biochem. Biophys. Res. Commun. 2010, 400, 684–688. [Google Scholar] [CrossRef] [Green Version]
- Sevetson, B.; Taylor, S.; Pan, Y. Cbfa1/RUNX2 Directs Specific Expression of the Sclerosteosis Gene (SOST). J. Biol. Chem. 2004, 279, 13849–13858. [Google Scholar] [CrossRef] [Green Version]
- Shan, Y.; Wang, L.; Li, G.; Shen, G.; Zhang, P.; Xu, Y. Methylation of Bone SOST Impairs SP7, RUNX2, and ERα Transactivation in Patients with Postmenopausal Osteoporosis. Biochem. Cell Biol. 2019, 97, 369–374. [Google Scholar] [CrossRef] [Green Version]
- Delgado-Calle, J.; Arozamena, J.; Pérez-López, J.; Bolado-Carrancio, A.; Sañudo, C.; Agudo, G.; de la Vega, R.; Alonso, M.A.; Rodríguez-Rey, J.C.; Riancho, J.A. Role of BMPs in the Regulation of Sclerostin as Revealed by an Epigenetic Modifier of Human Bone Cells. Mol. Cell. Endocrinol. 2013, 369, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Kamiya, N. The Role of BMPs in Bone Anabolism and Their Potential Targets SOST and DKK1. Curr. Mol. Pharmacol. 2012, 5, 153–163. [Google Scholar] [CrossRef]
- Galea, G.L.; Price, J.S.; Lanyon, L.E. Estrogen Receptors’ Roles in the Control of Mechanically Adaptive Bone (re)modeling. Bonekey Rep. 2013, 2, 413. [Google Scholar] [CrossRef]
- Delgado-Calle, J.; Sañudo, C.; Bolado, A.; Fernández, A.F.; Arozamena, J.; Pascual-Carra, M.A.; Rodriguez-Rey, J.C.; Fraga, M.F.; Bonewald, L.; Riancho, J.A. DNA Methylation Contributes to the Regulation of Sclerostin Expression in Human Osteocytes. J. Bone Miner. Res. 2012, 27, 926–937. [Google Scholar] [CrossRef] [PubMed]
- Delgado-Calle, J.; Fernández, A.F.; Sainz, J.; Zarrabeitia, M.T.; Sañudo, C.; García-Renedo, R.; Pérez-Núñez, M.I.; García-Ibarbia, C.; Fraga, M.F.; Riancho, J.A. Genome-Wide Profiling of Bone Reveals Differentially Methylated Regions in Osteoporosis and Osteoarthritis. Arthritis Rheum. 2013, 65, 197–205. [Google Scholar] [CrossRef]
- Cohen-Kfir, E.; Artsi, H.; Levin, A.; Abramowitz, E.; Bajayo, A.; Gurt, I.; Zhong, L.; D’Urso, A.; Toiber, D.; Mostoslavsky, R.; et al. Sirt1 Is a Regulator of Bone Mass and a Repressor of Sost Encoding for Sclerostin, a Bone Formation Inhibitor. Endocrinology 2011, 152, 4514–4524. [Google Scholar] [CrossRef] [PubMed]
- Kramer, I.; Baertschi, S.; Halleux, C.; Keller, H.; Kneissel, M. Mef2c Deletion in Osteocytes Results in Increased Bone Mass. J. Bone Miner. Res. 2012, 27, 360–373. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.; Jiang, X.; Dai, Z.; Guo, X.; Weng, T.; Wang, J.; Li, Y.; Feng, G.; Gao, X.; He, L. Sclerostin Mediates Bone Response to Mechanical Unloading Through Antagonizing Wnt/β-Catenin Signaling. J. Bone Miner. Res. 2009, 24, 1651–1661. [Google Scholar] [CrossRef] [PubMed]
- Delgado-Calle, J.; Riancho, J.A.; Klein-Nulend, J. Nitric Oxide Is Involved in the down-Regulation of SOST Expression Induced by Mechanical Loading. Calcif. Tissue Int. 2014, 94, 414–422. [Google Scholar] [CrossRef]
- Galea, G.L.; Sunters, A.; Meakin, L.B.; Zaman, G.; Sugiyama, T.; Lanyon, L.E.; Price, J.S. Sost down-Regulation by Mechanical Strain in Human Osteoblastic Cells Involves PGE2 Signaling via EP4. FEBS Lett. 2011, 585, 2450–2454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonnet, N.; Standley, K.N.; Bianchi, E.N.; Stadelmann, V.; Foti, M.; Conway, S.J.; Ferrari, S.L. The Matricellular Protein Periostin Is Required for Sost Inhibition and the Anabolic Response to Mechanical Loading and Physical Activity. J. Biol. Chem. 2009, 284, 35939–35950. [Google Scholar] [CrossRef] [Green Version]
- Bonnet, N.; Conway, S.J.; Ferrari, S.L. Regulation of Β Catenin Signaling and Parathyroid Hormone Anabolic Effects in Bone by the Matricellular Protein Periostin. Proc. Natl. Acad. Sci. USA 2012, 109, 15048–15053. [Google Scholar] [CrossRef] [Green Version]
- Koide, M.; Kobayashi, Y. Regulatory Mechanisms of Sclerostin Expression during Bone Remodeling. J. Bone Miner. Res. 2019, 37, 9–17. [Google Scholar] [CrossRef]
- Ukita, M.; Yamaguchi, T.; Ohata, N.; Tamura, M. Sclerostin Enhances Adipocyte Differentiation in 3T3-L1 Cells. J. Cell. Biochem. 2016, 117, 1419–1428. [Google Scholar] [CrossRef] [Green Version]
- You, L.; Chen, L.; Pan, L.; Peng, Y.; Chen, J. SOST Gene Inhibits Osteogenesis from Adipose-Derived Mesenchymal Stem Cells by Inducing Th17 Cell Differentiation. Cell. Physiol. Biochem. 2018, 48, 1030–1040. [Google Scholar] [CrossRef]
- Kim, S.P.; Frey, J.L.; Li, Z.; Kushwaha, P.; Zoch, M.L.; Tomlinson, R.E.; Da, H.; Aja, S.; Noh, H.L.; Kim, J.K.; et al. Sclerostin Influences Body Composition by Regulating Catabolic and Anabolic Metabolism in Adipocytes. Proc. Natl. Acad. Sci. USA 2017, 114, E11238–E11247. [Google Scholar] [CrossRef] [Green Version]
- Cosman, F.; Crittenden, D.B.; Adachi, J.D.; Binkley, N.; Czerwinski, E.; Ferrari, S.; Hofbauer, L.C.; Lau, E.; Lewiecki, E.M.; Miyauchi, A.; et al. Romosozumab Treatment in Postmenopausal Women with Osteoporosis. N. Engl. J. Med. 2016, 375, 1532–1543. [Google Scholar] [CrossRef]
- McClung, M.R.; Grauer, A.; Boonen, S.; Bolognese, M.A.; Brown, J.P.; Diez-Perez, A.; Langdahl, B.L.; Reginster, J.-Y.; Zanchetta, J.R.; Wasserman, S.M.; et al. Romosozumab in Postmenopausal Women with Low Bone Mineral Density. N. Engl. J. Med. 2014, 370, 412–420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Recker, R.R.; Benson, C.T.; Matsumoto, T.; Bolognese, M.A.; Robins, D.A.; Alam, J.; Chiang, A.Y.; Hu, L.; Krege, J.H.; Sowa, H.; et al. A Randomized, Double-Blind Phase 2 Clinical Trial of Blosozumab, a Sclerostin Antibody, in Postmenopausal Women with Low Bone Mineral Density. J. Bone Miner. Res. 2015, 30, 216–224. [Google Scholar] [CrossRef] [PubMed]
- Langdahl, B.L.; Libanati, C.; Crittenden, D.B.; Bolognese, M.A.; Brown, J.P.; Daizadeh, N.S.; Dokoupilova, E.; Engelke, K.; Finkelstein, J.S.; Genant, H.K.; et al. Romosozumab (sclerostin Monoclonal Antibody) versus Teriparatide in Postmenopausal Women with Osteoporosis Transitioning from Oral Bisphosphonate Therapy: A Randomised, Open-Label, Phase 3 Trial. Lancet 2017, 390, 1585–1594. [Google Scholar] [CrossRef]
- Glorieux, F.H.; Devogelaer, J.-P.; Durigova, M.; Goemaere, S.; Hemsley, S.; Jakob, F.; Junker, U.; Ruckle, J.; Seefried, L.; Winkle, P.J. BPS804 Anti-Sclerostin Antibody in Adults With Moderate Osteogenesis Imperfecta: Results of a Randomized Phase 2a Trial. J. Bone Miner. Res. 2017, 32, 1496–1504. [Google Scholar] [CrossRef]
- Lewiecki, E.M.; Blicharski, T.; Goemaere, S.; Lippuner, K.; Meisner, P.D.; Miller, P.D.; Miyauchi, A.; Maddox, J.; Chen, L.; Horlait, S. A Phase III Randomized Placebo-Controlled Trial to Evaluate Efficacy and Safety of Romosozumab in Men With Osteoporosis. J. Clin. Endocrinol. Metab. 2018, 103, 3183–3193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Ominsky, M.S.; Warmington, K.S.; Morony, S.; Gong, J.; Cao, J.; Gao, Y.; Shalhoub, V.; Tipton, B.; Haldankar, R.; et al. Sclerostin Antibody Treatment Increases Bone Formation, Bone Mass, and Bone Strength in a Rat Model of Postmenopausal Osteoporosis. J. Bone Miner. Res. 2009, 24, 578–588. [Google Scholar] [CrossRef]
- Ominsky, M.S.; Vlasseros, F.; Jolette, J.; Smith, S.Y.; Stouch, B.; Doellgast, G.; Gong, J.; Gao, Y.; Cao, J.; Graham, K.; et al. Two Doses of Sclerostin Antibody in Cynomolgus Monkeys Increases Bone Formation, Bone Mineral Density, and Bone Strength. J. Bone Miner. Res. 2010, 25, 948–959. [Google Scholar] [CrossRef]
- Stolina, M.; Dwyer, D.; Niu, Q.-T.; Villasenor, K.S.; Kurimoto, P.; Grisanti, M.; Han, C.-Y.; Liu, M.; Li, X.; Ominsky, M.S.; et al. Temporal Changes in Systemic and Local Expression of Bone Turnover Markers during Six Months of Sclerostin Antibody Administration to Ovariectomized Rats. Bone 2014, 67, 305–313. [Google Scholar] [CrossRef]
- Markham, A. Romosozumab: First Global Approval. Drugs 2019, 79, 471–476. [Google Scholar] [CrossRef] [PubMed]
- Ohazama, A.; Porntaveetus, T.; Ota, M.S.; Herz, J.; Sharpe, P.T. Lrp4: A Novel Modulator of Extracellular Signaling in Craniofacial Organogenesis. Am. J. Med. Genet. A 2010, 152A, 2974–2983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blish, K.R.; Wang, W.; Willingham, M.C.; Du, W.; Birse, C.E.; Krishnan, S.R.; Brown, J.C.; Hawkins, G.A.; Garvin, A.J.; D’Agostino, R.B., Jr.; et al. A Human Bone Morphogenetic Protein Antagonist Is down-Regulated in Renal Cancer. Mol. Biol. Cell 2008, 19, 457–464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beaudoin, G.M.J., 3rd; Sisk, J.M.; Coulombe, P.A.; Thompson, C.C. Hairless Triggers Reactivation of Hair Growth by Promoting Wnt Signaling. Proc. Natl. Acad. Sci. USA 2005, 102, 14653–14658. [Google Scholar] [CrossRef] [Green Version]
- Ahn, Y.; Sims, C.; Logue, J.M.; Weatherbee, S.D.; Krumlauf, R. Lrp4 and Wise Interplay Controls the Formation and Patterning of Mammary and Other Skin Appendage Placodes by Modulating Wnt Signaling. Development 2013, 140, 583–593. [Google Scholar] [CrossRef] [Green Version]
- Guidato, S.; Itasaki, N. Wise Retained in the Endoplasmic Reticulum Inhibits Wnt Signaling by Reducing Cell Surface LRP6. Dev. Biol. 2007, 310, 250–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kassai, Y.; Munne, P.; Hotta, Y.; Penttilä, E.; Kavanagh, K.; Ohbayashi, N.; Takada, S.; Thesleff, I.; Jernvall, J.; Itoh, N. Regulation of Mammalian Tooth Cusp Patterning by Ectodin. Science 2005, 309, 2067–2070. [Google Scholar] [CrossRef] [Green Version]
- Munne, P.M.; Tummers, M.; Jarvinen, E.; Thesleff, I.; Jernvall, J. Tinkering with the Inductive Mesenchyme: Sostdc1 Uncovers the Role of Dental Mesenchyme in Limiting Tooth Induction. Development 2009, 136, 393–402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahn, Y.; Sanderson, B.W.; Klein, O.D.; Krumlauf, R. Inhibition of Wnt Signaling by Wise (Sostdc1) and Negative Feedback from Shh Controls Tooth Number and Patterning. Development 2010, 137, 3221–3231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, S.E.; Jomary, C. Secreted Frizzled-Related Proteins: Searching for Relationships and Patterns. Bioessays 2002, 24, 811–820. [Google Scholar] [CrossRef]
- Bovolenta, P.; Esteve, P.; Ruiz, J.M.; Cisneros, E.; Lopez-Rios, J. Beyond Wnt Inhibition: New Functions of Secreted Frizzled-Related Proteins in Development and Disease. J. Cell Sci. 2008, 121, 737–746. [Google Scholar] [CrossRef] [Green Version]
- Bafico, A.; Gazit, A.; Pramila, T.; Finch, P.W.; Yaniv, A.; Aaronson, S.A. Interaction of Frizzled Related Protein (FRP) with Wnt Ligands and the Frizzled Receptor Suggests Alternative Mechanisms for FRP Inhibition of Wnt Signaling. J. Biol. Chem. 1999, 274, 16180–16187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez, J.; Esteve, P.; Weinl, C.; Ruiz, J.M.; Fermin, Y.; Trousse, F.; Dwivedy, A.; Holt, C.; Bovolenta, P. SFRP1 Regulates the Growth of Retinal Ganglion Cell Axons through the Fz2 Receptor. Nat. Neurosci. 2005, 8, 1301–1309. [Google Scholar] [CrossRef]
- Cho, S.W.; Her, S.J.; Sun, H.J.; Choi, O.K.; Yang, J.-Y.; Kim, S.W.; Kim, S.Y.; Shin, C.S. Differential Effects of Secreted Frizzled-Related Proteins (SFRPs) on Osteoblastic Differentiation of Mouse Mesenchymal Cells and Apoptosis of Osteoblasts. Biochem. Biophys. Res. Commun. 2008, 367, 399–405. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.K.; Samos, C.H.; Peoples, R.; Pérez-Jurado, L.A.; Nusse, R.; Francke, U. A Novel Human Homologue of the Drosophila Frizzled Wnt Receptor Gene Binds Wingless Protein and Is in the Williams Syndrome Deletion at 7q11.23. Hum. Mol. Genet. 1997, 6, 465–472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, K.; Wang, S.; Julius, M.A.; Kitajewski, J.; Moos, M.; Luyten, F.P. The Cysteine-Rich Frizzled Domain of Frzb-1 Is Required and Sufficient for Modulation of Wnt Signaling. Proc. Natl. Acad. Sci. USA 1997, 94, 11196–11200. [Google Scholar] [CrossRef] [Green Version]
- Lopez-Rios, J.; Esteve, P.; Ruiz, J.M.; Bovolenta, P. The Netrin-Related Domain of SFRP1 Interacts with Wnt Ligands and Antagonizes Their Activity in the Anterior Neural Plate. Neural Dev. 2008, 3, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uren, A.; Reichsman, F.; Anest, V.; Taylor, W.G.; Muraiso, K.; Bottaro, D.P.; Cumberledge, S.; Rubin, J.S. Secreted Frizzled-Related Protein-1 Binds Directly to Wingless and Is a Biphasic Modulator of Wnt Signaling. J. Biol. Chem. 2000, 275, 4374–4382. [Google Scholar] [CrossRef] [Green Version]
- Von Marschall, Z.; Fisher, L.W. Secreted Frizzled-Related Protein-2 (SFRP2) Augments Canonical Wnt3a-Induced Signaling. Biochem. Biophys. Res. Commun. 2010, 400, 299–304. [Google Scholar] [CrossRef] [Green Version]
- Chung, Y.-S.; Baylink, D.J.; Srivastava, A.K.; Amaar, Y.; Tapia, B.; Kasukawa, Y.; Mohan, S. Effects of Secreted Frizzled-Related Protein 3 on Osteoblasts in vitro. J. Bone Miner. Res. 2004, 19, 1395–1402. [Google Scholar] [CrossRef]
- Yoshino, K.; Rubin, J.S.; Higinbotham, K.G.; Uren, A.; Anest, V.; Plisov, S.Y.; Perantoni, A.O. Secreted Frizzled-Related Proteins Can Regulate Metanephric Development. Mech. Dev. 2001, 102, 45–55. [Google Scholar] [CrossRef]
- Mii, Y.; Taira, M. Secreted Frizzled-Related Proteins Enhance the Diffusion of Wnt Ligands and Expand Their Signalling Range. Development 2009, 136, 4083–4088. [Google Scholar] [CrossRef] [Green Version]
- Alfaro, M.P.; Vincent, A.; Saraswati, S.; Thorne, C.A.; Hong, C.C.; Lee, E.; Young, P.P. SFRP2 Suppression of Bone Morphogenic Protein (BMP) and Wnt Signaling Mediates Mesenchymal Stem Cell (MSC) Self-Renewal Promoting Engraftment and Myocardial Repair. J. Biol. Chem. 2010, 285, 35645–35653. [Google Scholar] [CrossRef] [Green Version]
- Häusler, K.D.; Horwood, N.J.; Chuman, Y.; Fisher, J.L.; Ellis, J.; Martin, T.J.; Rubin, J.S.; Gillespie, M.T. Secreted Frizzled-Related Protein-1 Inhibits RANKL-Dependent Osteoclast Formation. J. Bone Miner. Res. 2004, 19, 1873–1881. [Google Scholar] [CrossRef] [PubMed]
- Gaur, T.; Wixted, J.J.; Hussain, S.; O’Connell, S.L.; Morgan, E.F.; Ayers, D.C.; Komm, B.S.; Bodine, P.V.; Stein, G.S.; Lian, J.B. Secreted Frizzled Related Protein 1 Is a Target to Improve Fracture Healing. J. Cell. Physiol. 2009, 220, 174–181. [Google Scholar] [CrossRef] [Green Version]
- Vaes, B.L.T.; Dechering, K.J.; van Someren, E.P.; Hendriks, J.M.A.; van de Ven, C.J.J.M.; Feijen, A.; Mummery, C.L.; Reinders, M.J.T.; Olijve, W.; van Zoelen, E.J.J.; et al. Microarray Analysis Reveals Expression Regulation of Wnt Antagonists in Differentiating Osteoblasts. Bone 2005, 36, 803–811. [Google Scholar] [CrossRef]
- Bravo, D.; Salduz, A.; Shogren, K.L.; Okuno, M.N.; Herrick, J.L.; Okuno, S.H.; Galindo, M.; van Wijnen, A.J.; Yaszemski, M.J.; Maran, A. Decreased Local and Systemic Levels of SFRP3 Protein in Osteosarcoma Patients. Gene 2018, 674, 1–7. [Google Scholar] [CrossRef]
- Kristensen, I.B.; Christensen, J.H.; Lyng, M.B.; Møller, M.B.; Pedersen, L.; Rasmussen, L.M.; Ditzel, H.J.; Abildgaard, N. Expression of Osteoblast and Osteoclast Regulatory Genes in the Bone Marrow Microenvironment in Multiple Myeloma: Only up-Regulation of Wnt Inhibitors SFRP3 and DKK1 Is Associated with Lytic Bone Disease. Leuk. Lymphoma 2014, 55, 911–919. [Google Scholar] [CrossRef]
- Mori, K.; Kitazawa, R.; Kondo, T.; Mori, M.; Hamada, Y.; Nishida, M.; Minami, Y.; Haraguchi, R.; Takahashi, Y.; Kitazawa, S. Diabetic Osteopenia by Decreased β-Catenin Signaling Is Partly Induced by Epigenetic Derepression of SFRP-4 Gene. PLoS ONE 2014, 9, e102797. [Google Scholar] [CrossRef]
- Berndt, T.; Kumar, R. Phosphatonins and the Regulation of Phosphate Homeostasis. Annu. Rev. Physiol. 2007, 69, 341–359. [Google Scholar] [CrossRef] [PubMed]
- Bodine, P.V.N.; Billiard, J.; Moran, R.A.; Ponce-de-Leon, H.; McLarney, S.; Mangine, A.; Scrimo, M.J.; Bhat, R.A.; Stauffer, B.; Green, J.; et al. The Wnt Antagonist Secreted Frizzled-Related Protein-1 Controls Osteoblast and Osteocyte Apoptosis. J. Cell. Biochem. 2005, 96, 1212–1230. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, J.C.; Kodjabachian, L.; Rebbert, M.L.; Rattner, A.; Smallwood, P.M.; Samos, C.H.; Nusse, R.; Dawid, I.B.; Nathans, J. A New Secreted Protein That Binds to Wnt Proteins and Inhibits Their Activities. Nature 1999, 398, 431–436. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.W.; Yang, J.-Y.; Sun, H.J.; Jung, J.Y.; Her, S.J.; Cho, H.Y.; Choi, H.J.; Kim, S.W.; Kim, S.Y.; Shin, C.S. Wnt Inhibitory Factor (WIF)-1 Inhibits Osteoblastic Differentiation in Mouse Embryonic Mesenchymal Cells. Bone 2009, 44, 1069–1077. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Chen, C.; Liu, H.; Liu, X.; Zhao, H.; Hu, J. Gossypol Promotes Wnt/β-Catenin Signaling through WIF1 in Ovariectomy-Induced Osteoporosis. BioMed Res. Int. 2019, 2019, 1–10. [Google Scholar] [CrossRef]
- Leanza, G.; Fontana, F.; Lee, S.-Y.; Remedi, M.S.; Schott, C.; Ferron, M.; Hamilton-Hall, M.; Alippe, Y.; Strollo, R.; Napoli, N.; et al. Gain-of-Function Lrp5 Mutation Improves Bone Mass and Strength and Delays Hyperglycemia in a Mouse Model of Insulin-Deficient Diabetes. J. Bone Miner. Res. 2021, 36, 1403–1415. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Wu, D.; Sun, X.; Fan, Y.; Zha, R.; Jalali, A.; Feng, Y.; Li, K.; Sano, T.; Vike, N.; et al. Overexpression of Lrp5 enhanced the anti-breast cancer effects of osteocytes in bone. Bone Res. 2021, 9, 32. [Google Scholar] [CrossRef]





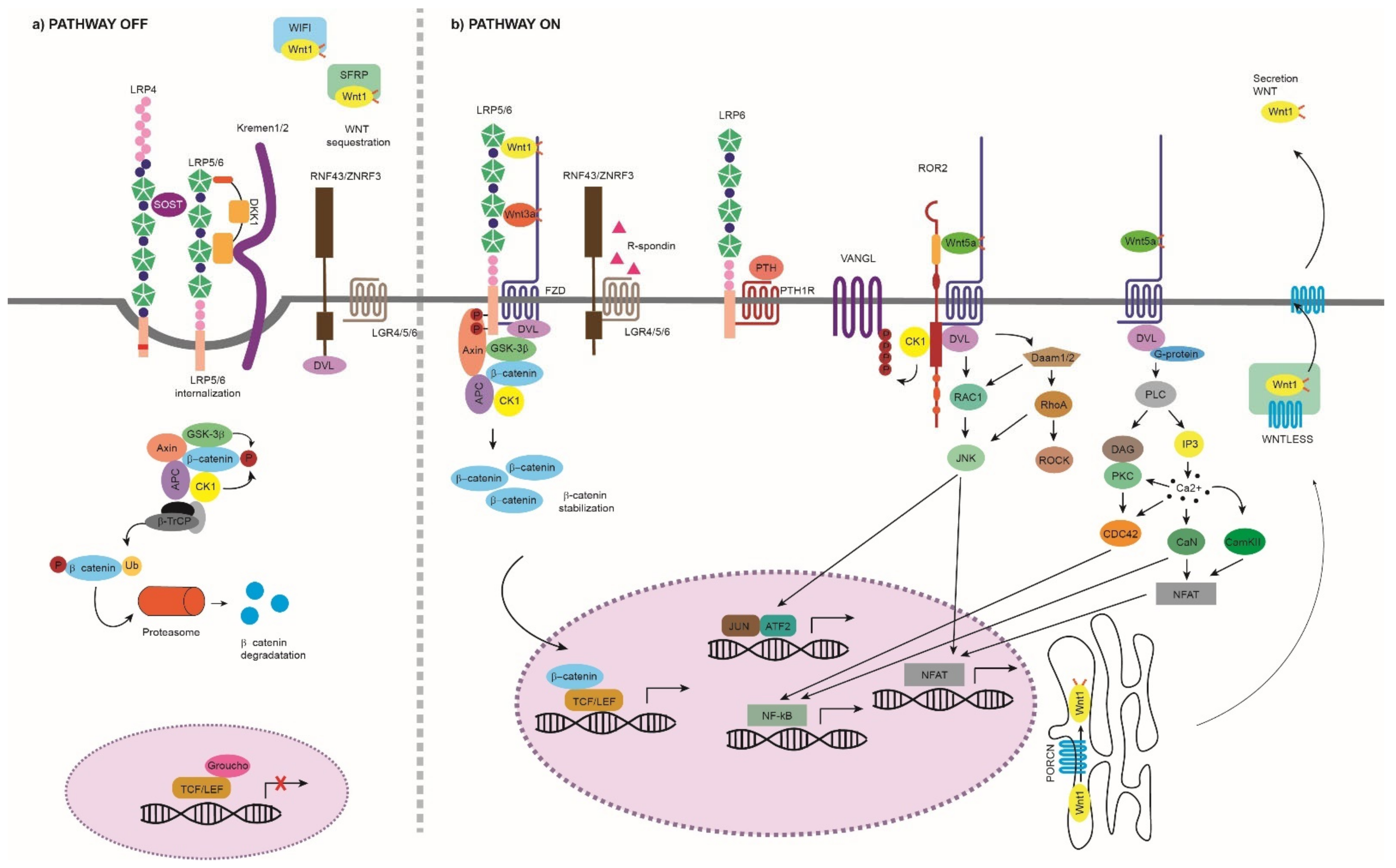{kind=link}
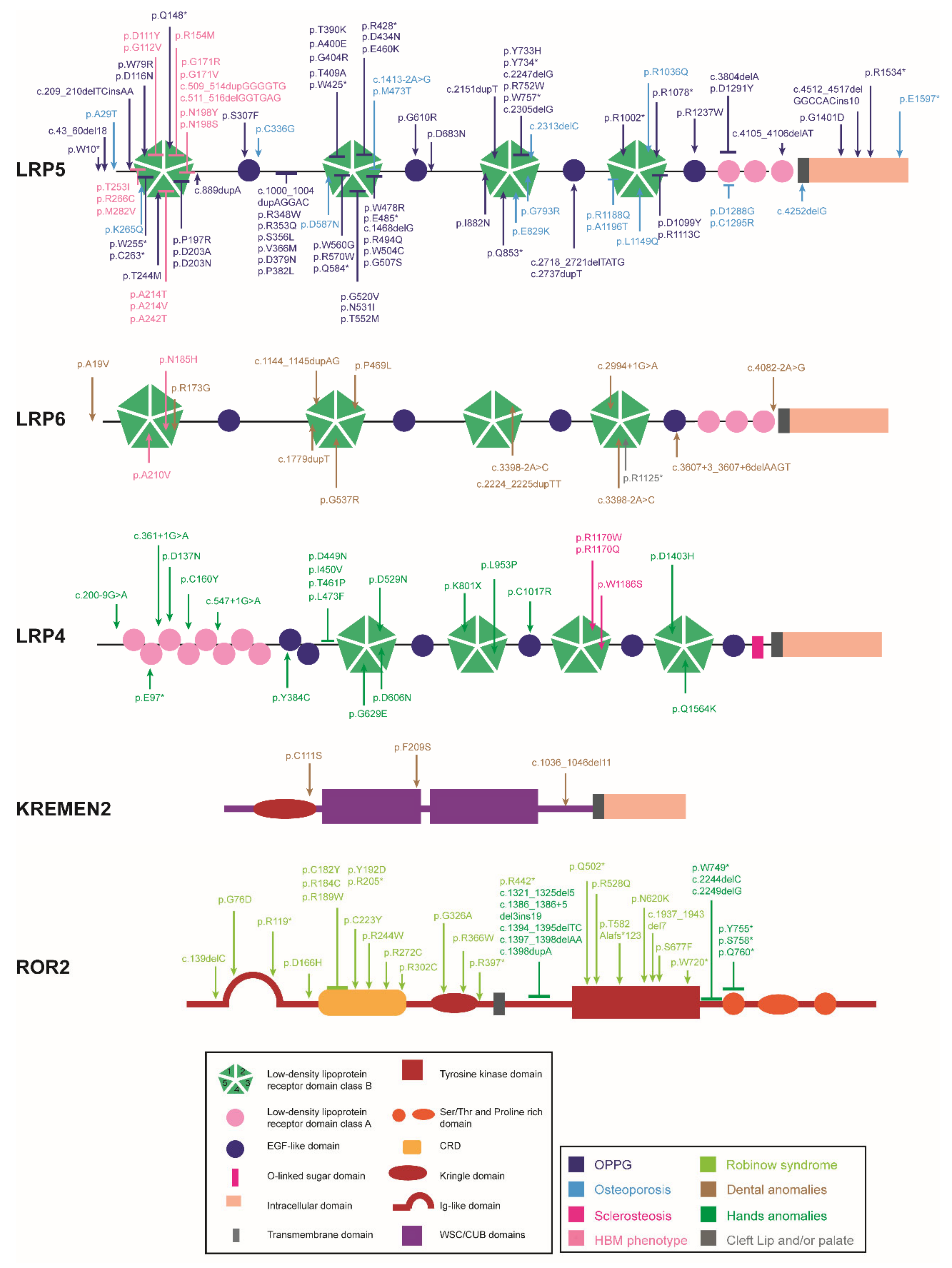{kind=link}
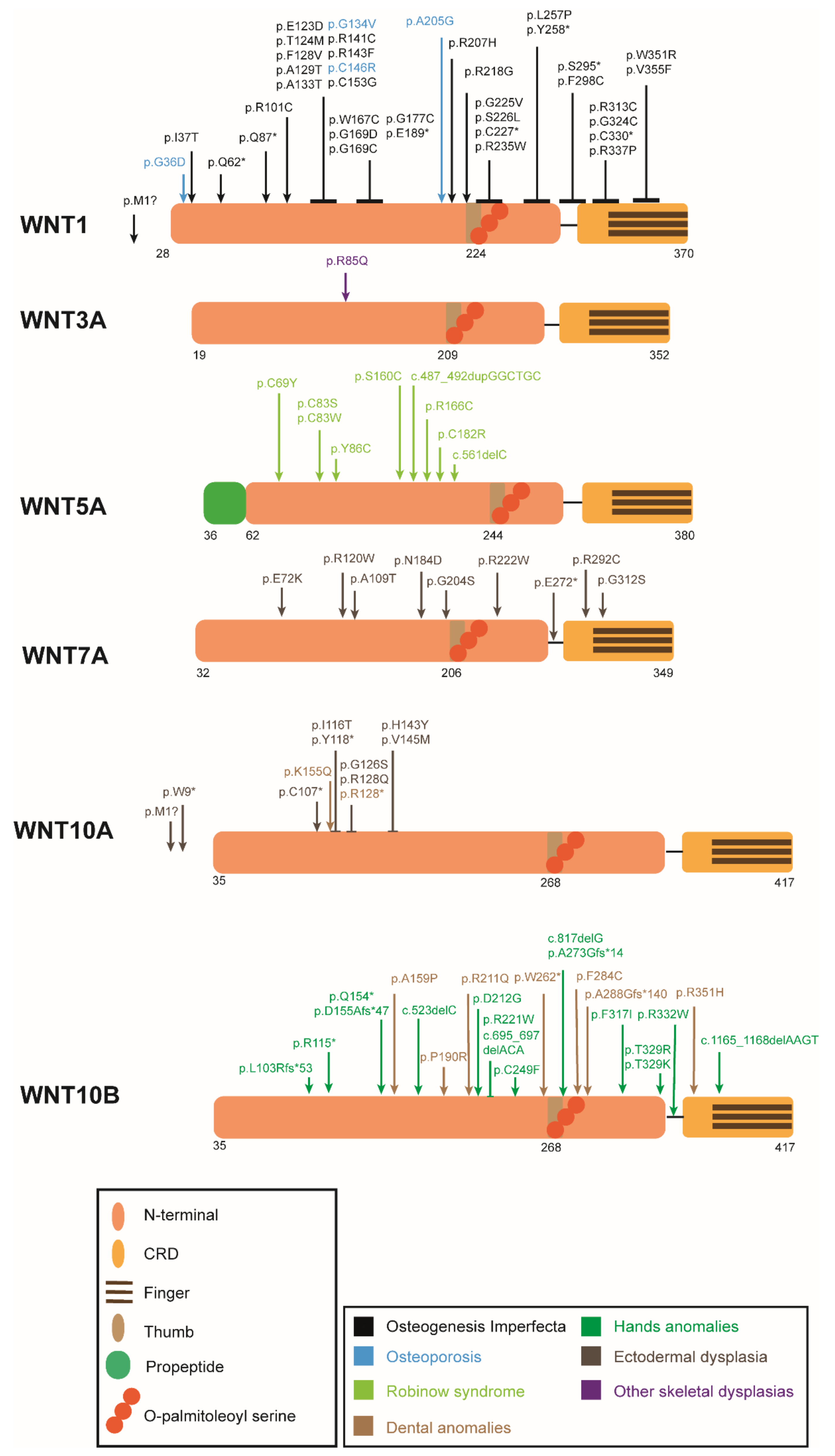{kind=link}
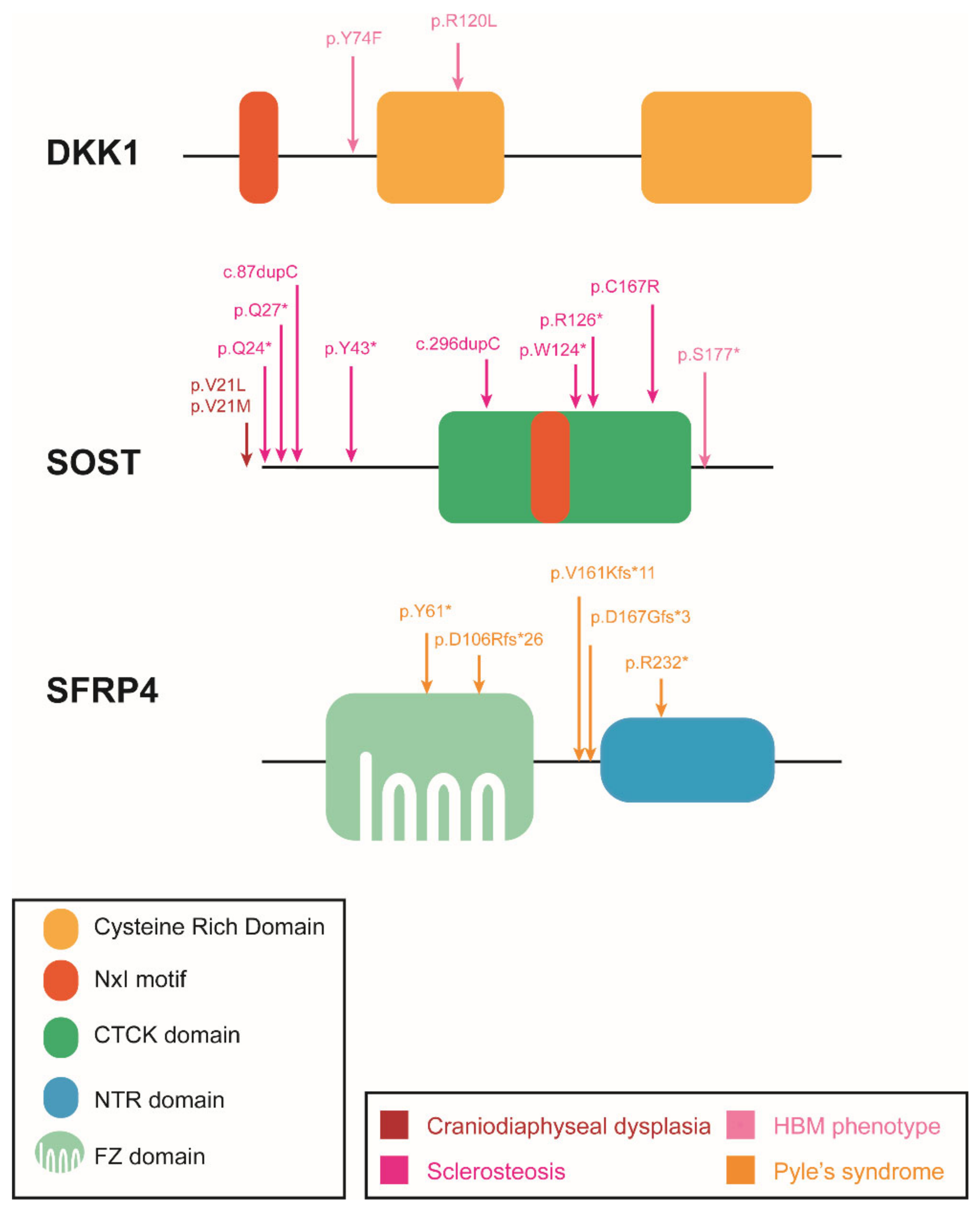{kind=link}
| Disease/Trait | OMIM | Phenotype | Gene | Inh. | Comment | Refs. |
|---|---|---|---|---|---|---|
| Osteogenesis imperfecta XV | 615220 |
| WNT1 | AR/AD | LoF | [35,36,37] |
| Osteoporosis-pseudoglioma syndrome (OPPG) | 259770 |
| LRP5 | AR | LoF | [38,39] |
| Osteoporosis | 615221 |
| WNT1 | AD | LoF | [35] |
| 166710 | LRP5 | AD | LoF | [40,41,42] | ||
| Craniodiaphyseal dysplasia | 122860 |
| SOST | AD | LoF/DN | [43] |
| Sclerosteosis | 269500 |
| SOST | AR | LOF | [44,45,46] |
| 614305 | LRP4 | AD/AR | LOF | [47,48] | ||
| van Buchem disease | 239100 |
| SOST * | AR | * 52 kb downstream deletion | [49] |
| HBM phenotype 1 | 144750 607634 607636 |
| LRP5 | AD | GoF | [50,51,52] |
| LRP6 | AD | GoF | [53] | |||
| SOST | AR | LoF | [54] | |||
| DKK1 | AD | LoF | [55,56] | |||
| Robinow syndrome | 268310 |
| ROR2 | AR | LoF | [57,58,59] |
| 180700 | WNT5A | AD | LoF | [60] | ||
| Pyle’s syndrome | 265900 |
| SFRP4 | AR | LoF | [61] |
| Selective tooth agenesis, types 7, 4 and 8 | 616724 |
| LRP6 | AD | LoF | [62,63] |
| 150400 | WNT10A | AD/AR | LoF | [64,65] | ||
| 617073 | WNT10B | AD | LoF | [66] | ||
| Hand/foot anomalies: | ||||||
| Cenani–Lenz syndactyly syndrome | 212780 |
| LRP4 | AR | LoF | [67] |
| Isolated bilateral syndactyly |
| LRP4 | AR | LoF | [68,69] | |
| Brachydactyly type B1 | 113000 |
| ROR2 | AD | GoF | [70,71] |
| Split hand/foot malformation, isolated form, type 6 | 225300 |
| WNT10B | AR | LoF | [72] |
| Ectodermal dysplasia: | ||||||
| Ectodermal dysplasia 13 | 617392 |
| KRM1 | AR | [73,74] | |
| Al-Awadi-Raas-Rothschild syndrome (AARRS) | 276820 |
| WNT7A | AR | complete LOF | [75] |
| Furhman syndrome | 228930 |
| WNT7A | AR | partial LOF | [75] |
| Santos syndrome | 613005 |
| WNT7A | LoF | [76] | |
| Schöpf–Schulz–Passarge syndrome | 224750 |
| WNT10A | AR | LoF | [77] |
| Odonto-onycho-dermal dysplasia (OODD) | 257980 |
| WNT10A | AD/AR | LoF | [78] |
| Skeletal dysplasias: | ||||||
| Other skeletal dysplasias |
| WNT3A | AR | [79] | ||
| Gene | Model | Tissue Expression | Phenotype | Comments | Refs. | ||
|---|---|---|---|---|---|---|---|
| BM | BF | BR | |||||
| Lrp5 | KO | Total KO | ↓ | ↓ | ↓/= I | I ↓ in [80]; =in [81,82] | [80,81,82,83,84,85,86] |
| cKO-Dermo1-Cre | EM | = | = | - | [86] | ||
| cKO-Ocn-Cre | mature OB | ↓ | ↓ | = | [87] | ||
| cKO-α1(I)-Col-Cre | OB | = | = | - | [82] | ||
| cKO Rat 3.6Col1a1-Cre | OB | ↓ | - | - | [84] | ||
| cKO-Dmp1-Cre | OCy | ↓ | - | - | [88] | ||
| cKO-Vil1-Cre | ISC | ↓/= II | ↓/= II | - | II ↓ in [82]; =in [88] | [82,88] | |
| cKO-Col21-Cre | CD and OB progenitors | ↓ | - | - | [89] | ||
| KI-A214V or G171V | Total KI | ↑ | ↑ | = | [88] | ||
| cKI-Dmp1-Cre A214V or G171V | OCy | ↑ | ↑ | = | [88] | ||
| cKI-Prrx1-Cre A214V or G171V | MSC | ↑ | ↑ | = | [88] | ||
| cKI-Vil1-Cre A214V | ISC | = | = | = | [88] | ||
| cKI-Vil1-Cre G171V | ISC | ↑/= III | ↑/= III | = | III↑ in [82]; =in [88] | [82,88] | |
| cKI-1(I)-Col-Cre G171V | OB | = | = | = | [82] | ||
| Tg Rat3.6Col1a1G171V | OB | ↑ | [90] | ||||
| cKI-Ctsk-Cre A214V or G171V | mature OC | ↑ | ↑ | ↓ IV | Offtargets in Ocy line IV Only in female mice | [91] | |
| Lrp6 | KO | Total KO | - | - | - | NV | [83,86,92] |
| cKO-Dermo1-Cre | EM | = | = | = | [86] | ||
| Ringelschwanz | ↓ | = | ↑ | p.R868W mutation | [93,94] | ||
| cKO-Ocn-Cre | mature OB | ↓ | ↓ | = | [87] | ||
| cKO-Col2a1-Cre | CD and OB progenitors | ↓ | - | - | [89] | ||
| Lrp5 and Lrp6 | KO-Lrp5 and Het KO-Lrp6 | Total KO | ↓ | - | - | Severe limb abnormalities | [83] |
| cKO-Dermo-Cre | EM | ↓ | ↓ | - | NV | [86] | |
| cKO-Ocn-Cre | mature OB | ↓ | ↓ | = | NV | [87] | |
| cKO-Col2a1-Cre | CD and OB progenitors | ↓ | - | - | NV | [89] | |
| cKO-Rank-Cre | OC precursors | ↓ | ↓ | ↓ | [95] | ||
| cKO-Ctsk-Cre | mature OC | = | = | = | [95] | ||
| Lrp4 | KO | Total KO | NV | [96,97,98] | |||
| ECD | ↓ | ↑ | ↑ | Only ECD domain | [99] | ||
| cKO-OCN-Cre | mature OB | ↑ | ↑ | ↓ | [96,100] | ||
| cKO-Dermo1-Cre | OCy | ↑ | ↑ | ↓ | [100] | ||
| cKO-LysM-Cre | Myeloid cells | = | = | = | [96] | ||
| KI-Lrp4-R1170W | Total KI | ↑ | ↑ | = | [101] | ||
| KI-Lrp4-R1170Q | Total KI | ↑ | ↑ | = | [102] | ||
| Ror2 | KO | Total KO | - | - | - | NV | [8,103,104,105] |
| Het KO | Het KO | ↑ | = | ↓ | [103] | ||
| cKO-Rank-Cre | OC | ↑ | = | ↓ | [103] | ||
| cKO-Ctsk-Cre | mature OC | ↑ | = | ↓ | Only in trabecular bone | [103,106,107] | |
| Krm1 | KO | Total KO | = | = | - | [108] | |
| Krm2 | KO | Total KO | = | = | - | In young mice | [108] |
| KO | Total KO | ↑ | ↑ | - | In adult mice | [109] | |
| Tg Col1a1-Krm2 | OB | ↓ | ↓ | ↑ | [109] | ||
| Krm1 and Krm2 | KO | Total KO | ↑ | ↑ | = | [108] | |
| Lgr4 | KO | Total KO | ↓ | ↓/= V | ↑ | V ↓ [110,111]; = [112] | [110,111,112,113] |
| cKO-LysM | Myeloid cells | ↓ | = | ↑ | [112] | ||
| Lgr5 | KO | Total KO | NV | [114] | |||
| Lgr6 | KO | Total KO | = | = | = | [115,116] | |
| Gene | Model | Tissue Expression | Phenotype | Comments | Refs. | ||
|---|---|---|---|---|---|---|---|
| BM | BF | BR | |||||
| Wnt1 | KO | Total KO | - | - | - | NV | [207,208] |
| Het KO | Total Het KO | ↓ | = | = | Mild osteopenia | [207] | |
| Sway | Hypomorphic | ↓ | ↓ | = | G565del | [209,210] | |
| cKO-Prrx1-Cre | MSC | ↓ | ↓ | ↑ | [207] | ||
| cKO-Runx2-Cre | OB | ↓ | - | - | [211] | ||
| cKO-Dmp1-Cre | late OB and Ocy | ↓ | ↓ | - | [212] | ||
| cKO-Lyz2-Cre | Monocytic lineage (OC included) | = | - | - | [211] | ||
| Tg-Col1a1-tTA | OB, inducible | ↑ | ↑ | = | [211] | ||
| Dmp1-Cre-R26 het Wnt1 | Ov. in late OB and Ocy | ↑ | ↑ | = | HBM phenotype | [212] | |
| Wnt1 and Lrp5 | Tg-Wnt1; Lrp5-KO | OB-targeted inducible Ov. of Wnt1 in LRP5-KO | ↑ | ↑ | = | Similar phenotype to LRP5+/+ | [211] |
| Wnt3a | KO | Total KO | - | - | - | NV | [213,214] |
| Wnt4 | KO | Total KO | - | - | - | NV | [215] |
| cKO-Prrx1-Cre | MSC | ↑ I | - | - | I femoral, in females | [216] | |
| Tg-Col2.3kb-Wnt4 | OB | ↑ | ↑ | ↓ | [217] | ||
| Wnt4 and Wnt9a | dKO | Total dKO | ↓ | - | - | Ectopic cartilage formation | [218] |
| Wnt5a | KO | Total KO | - | - | - | NV | [219,220] |
| Het Wnt5a | Total Het KO | ↓ | ↓ | ↓ | Increased adipogenesis | [103,221] | |
| cKO-Osx-Cre | OB | ↓ | ↓ | ↓ | [103] | ||
| Wnt5b | KO | Total KO | - | - | - | [222] | |
| Wnt7a | KO | Total KO | - | - | - | Abnormal limb development | [223] |
| Wnt7b | KO | Total KO | - | - | - | NV | [224] |
| cKO-Dermo1-Cre | Skeletal progenitors | ↓ | - | - | [18] | ||
| Tg-OSX-Cre;R26 | Ov. in preOB | ↑ | ↑ | = | Little bone marrow space | [225] | |
| Tg-Col1-Cre;R26 | Ov. in mature OB | ↑ | ↑ | = | Little bone marrow space | [225] | |
| Col2-Cre; R26 | Ov. in OB and CD | ↑ | ↑ | - | Little bone marrow space | [210] | |
| Tg-Runx2-Wnt7b | Inducible total Ov. | ↑ | ↑ | = | [225] | ||
| Osx-rtTA; tetO-Cre; R26 | Inducible Ov. in OB | ↑ | ↑ | ↑ II | II less than formation | [226] | |
| Wnt9a | KO | Total KO | ↓ | - | - | NV, shortened long bones | [218] |
| Wnt9a and Ror1 | Ror1hyp/hyp; Wnt9a-KO | Total Wnt9a KO, hypomorphic Ror1 | ↓ | - | - | Deletion of Ror1 3rd exon, containing Ig-like domain | [166] |
| Wnt9a and Ror2 | dKO | Total dKO | ↓ | - | - | Shortened long bones | [166,218] |
| Wnt10a | KO | Total KO | ↓ | ↓ | - | Impaired dental formation | [227,228,229] |
| Wnt10b | Wnt10b-KO | Total KO | ↓ | ↓ | - | Early age dependent | [230,231,232] |
| Tg-FABP4-Wnt10b | Ov. in MSC | ↑ | - | - | Resistant to obesity and age/hormone- related bone loss | [232] | |
| Tg-Oc-Wnt10b | Ov. in OB | ↑ | ↑ | = | [231] | ||
| Tg-Oc-Wnt10b | Ov. in mature OB | ↑ | ↑ | ↑ | [230,231] | ||
| Wnt16 | KO | Total KO | ↓ | =/↓ III | ↑ | in cortical bone II I ↓ [233]; ↑ = [234] | [233,234,235,236] |
| cKO-Runx2-Cre | OB | ↓ | - | - | In cortical bone | [234] | |
| cKO-Dmp1-Cre | Ocy | = | - | - | [234] | ||
| CAG-Cre-ER | Tamoxifen-inducible | ↓ | ↓ | ↑ | In cortical bone | [237] | |
| Tg-2.3Kb rat-Col1a1-Wnt16 | OB | ↑ | =/↑ IV | =/↓ V | IV = [238]; ↑ [239] V = [238]; ↓ [239] | [238] | |
| Tg-Dmp1-Wnt16 | OCy | ↑ | ↑ | = | [240] | ||
| Gene | Model | Tissue Expression | Phenotype | Comments | Refs. | ||
|---|---|---|---|---|---|---|---|
| BM | BF | BR | |||||
| Dkk1 | KO | Total KO | - | - | - | NV | [306] |
| Het KO | Het KO | ↑ | ↑ | = | [307,308] | ||
| doubleridge | Hypomorphic | ↑ | - | - | Deletion of a regulatory region | [308,309] | |
| Rosa26-CreERT2 | Tamoxifen-inducible at 7 weeks | ↑ | ↑ | ↓ I | I Only the OC number | [310] | |
| cKO-OSX-Cre | Pre-OB | ↑ | ↑ | ↓ I | I Only the OC number | [310] | |
| cKO-Dmp1-Cre | OCy | ↑ | ↑ | ↓ I | I Only the OC number | [310,311] | |
| Tg-2.3 Kb rat-Col1a1 | OB | ↓ | ↓ | = | [312,313] | ||
| Wnt3 and Dkk1 | dKO | Total Het KO | ↑ | ↑ | = | NV | [314,315,316] |
| Dkk2 | KO | Total KO | ↓ | ↓ II | ↑ | Defects in mineralization; II Only BFR | [317] |
| Sost | KO | Total KO | ↑↑ | ↑ | = | [318,319] | |
| cKO-Prx2-Cre | Limb mesenchyme | ↑↑ | - | = | [320] | ||
| cKO-2.3KbCol1a1-Cre | OB | ↑ | - | = | [320] | ||
| Dmp1-Cre | OCy | ↑ | - | = | [320] | ||
| Col10a1-Cre | CD | ↑ | - | = | [320] | ||
| Tg-Dmp1-SOST | OCy | ↓ | ↓ | = | [321,322] | ||
| Tg-Oc-SOST | OB | ↓ | ↓ | = | [323] | ||
| Tg-SOST | Total Tg. | ↓ | ↓ | = | Insertion of ~158-kb from 3′-end of the DUSP3 to MEOX1 | [324] | |
| Sostdc 1 | KO | Total KO | ↑ | ↑ | ↓ | During the first 1.5 to 3 months of postnatal development | [325] |
| Sostdc1 and Lrp5 | dKO | Total KO | = | - | - | [325] | |
| Sfrp1 | KO | Total KO | ↑ | ↑ | = | Increased healing | [326,327,328] |
| Tg-Sfrp1 | Total Ov. | ↓ | - | - | Osteopenia | [329] | |
| Sfrp2 | KO | Total KO | = | = | = | Syndactyly | [326,330] |
| Sfrp4 | KO | Total KO | ↑I II/↓ IV | ↑ III | ↓ III | III in trabecular bone; IV in cortical bone | [61,106,331] |
| Tg-Col1a1-Sfrp4 | Ov. in OB | ↓ | ↓ | = | [332] | ||
| Sfrp5 | KO | Total KO | - | - | - | [333] | |
| Sfrp4 and Ror2 | Sfrp4-KO + Ror2-cKO | Total Sfrp4-KO; Ror2-cKO in OC | ↑ V | - | ↓ V | V in trabecular bone | [106] |
| Wif-1 | KO | Total KO | = | = | = | [334] | |
| Tg-2.3Col1a-Wif-1 | Ov. in mature OB | = | = | = | [335] | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martínez-Gil, N.; Ugartondo, N.; Grinberg, D.; Balcells, S. Wnt Pathway Extracellular Components and Their Essential Roles in Bone Homeostasis. Genes 2022, 13, 138. https://doi.org/10.3390/genes13010138
Martínez-Gil N, Ugartondo N, Grinberg D, Balcells S. Wnt Pathway Extracellular Components and Their Essential Roles in Bone Homeostasis. Genes. 2022; 13(1):138. https://doi.org/10.3390/genes13010138
Chicago/Turabian StyleMartínez-Gil, Núria, Nerea Ugartondo, Daniel Grinberg, and Susanna Balcells. 2022. "Wnt Pathway Extracellular Components and Their Essential Roles in Bone Homeostasis" Genes 13, no. 1: 138. https://doi.org/10.3390/genes13010138