
A Curious Novel Combination of Nucleophosmin (NPM1) Gene Mutations Leading to Aberrant Cytoplasmic Dislocation of NPM1 in Acute Myeloid Leukemia (AML)

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patients’ Samples
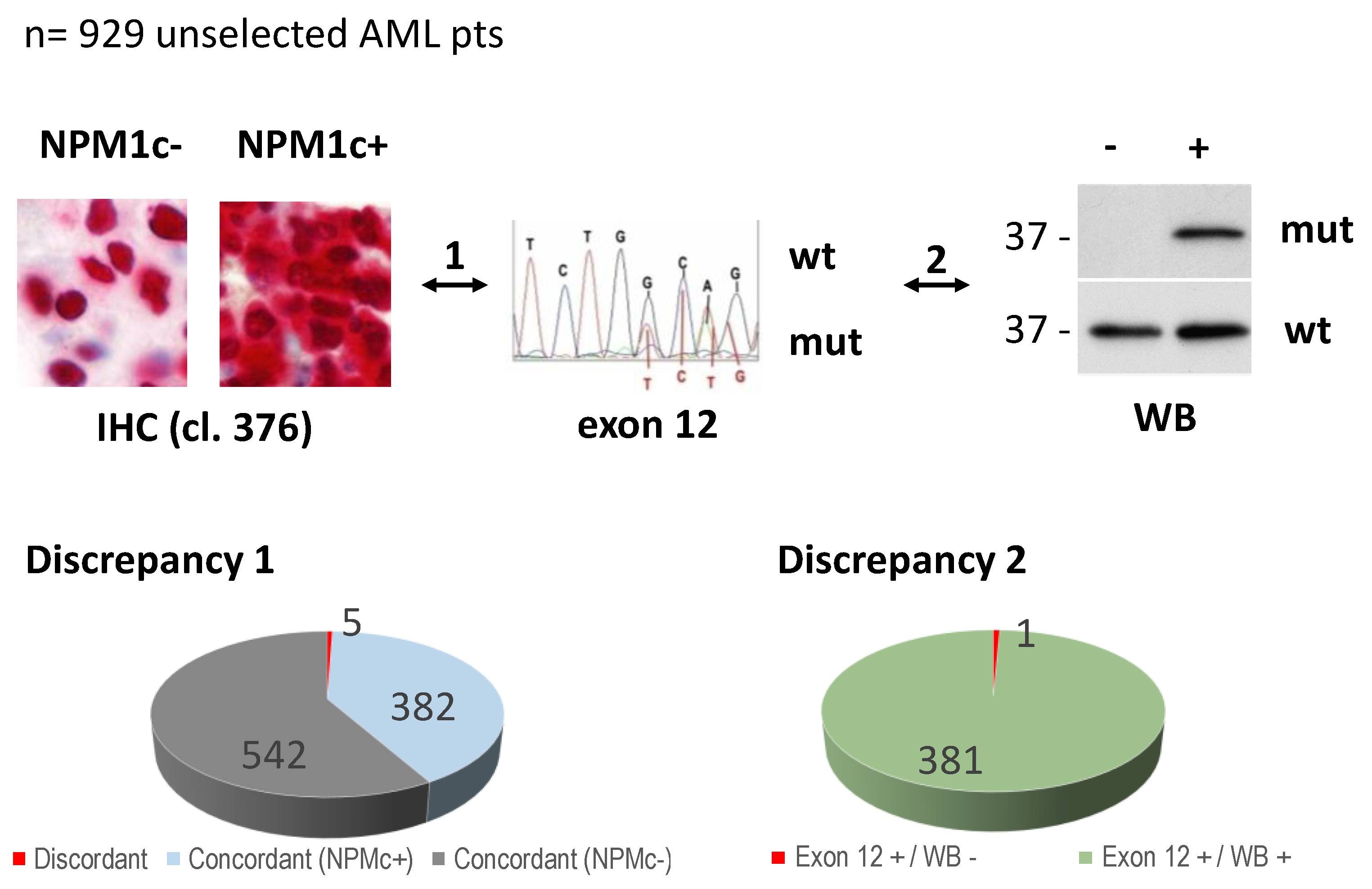
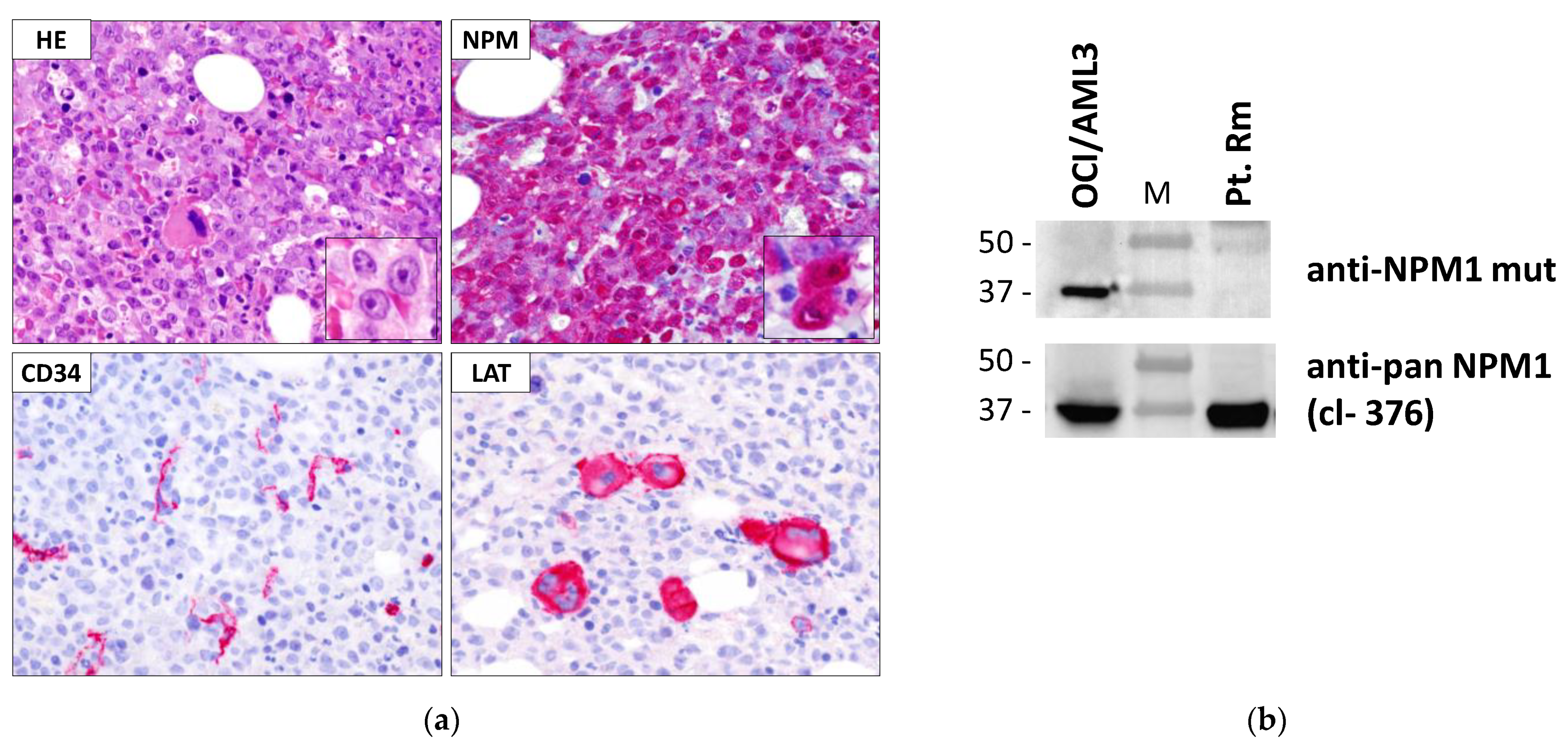
2.2. Immunohistochemical Analysis
2.3. Detection of NPM1 Mutant Protein by Western Blot Analysis
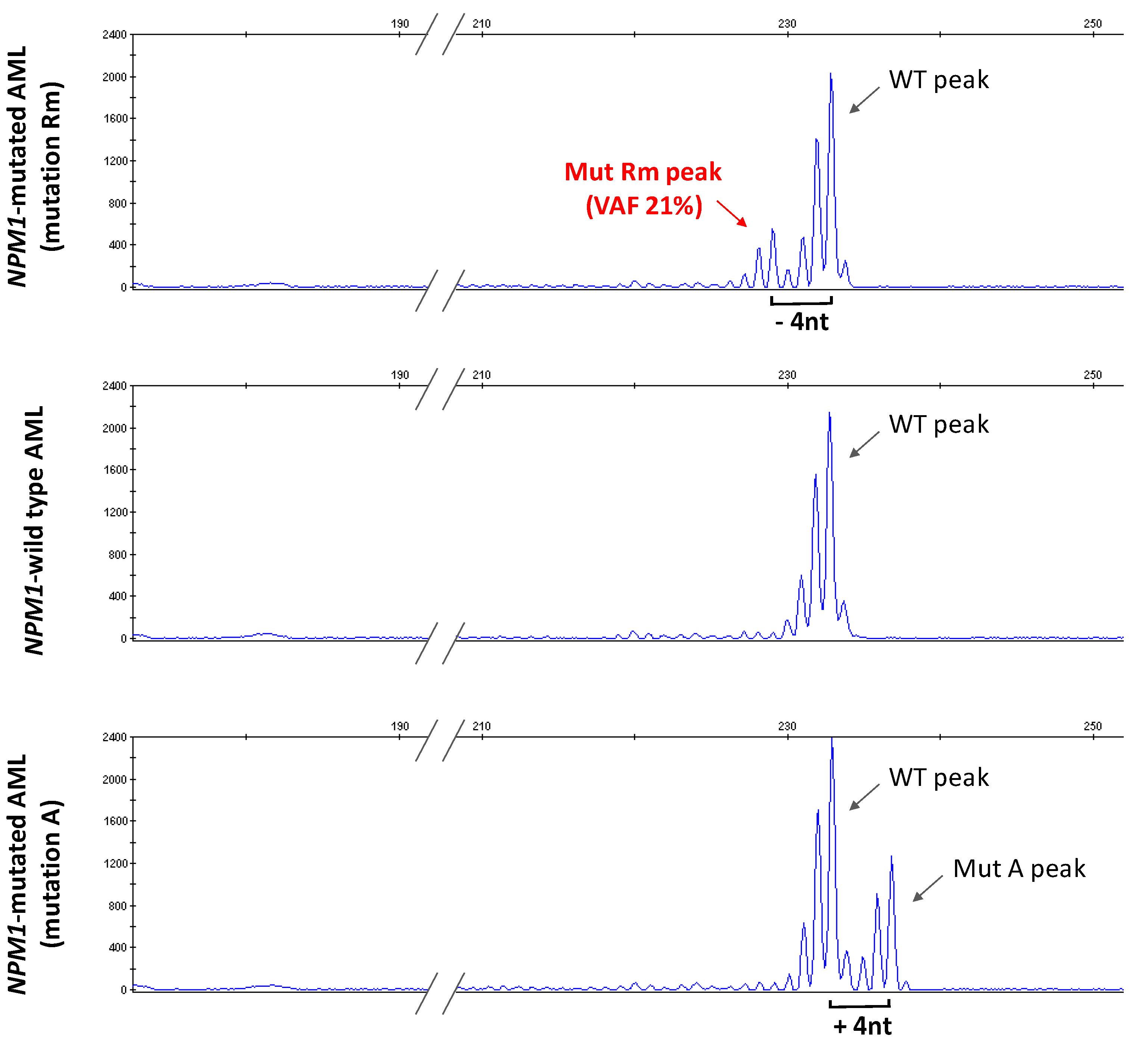
2.4. Sanger Sequencing of NPM1
2.5. Targeted Deep Sequencing
2.6. Genomic DNA NPM1 Fragment Analysis
2.7. pEGFP-C1-NPM1 Plasmid Constructs, Cell Transfection and Immunofluorescence Analysis
2.8. Real-Time Quantitative Polymerase Chain Reaction (RTq-PCR) for NPM1 Mutation A
3. Results
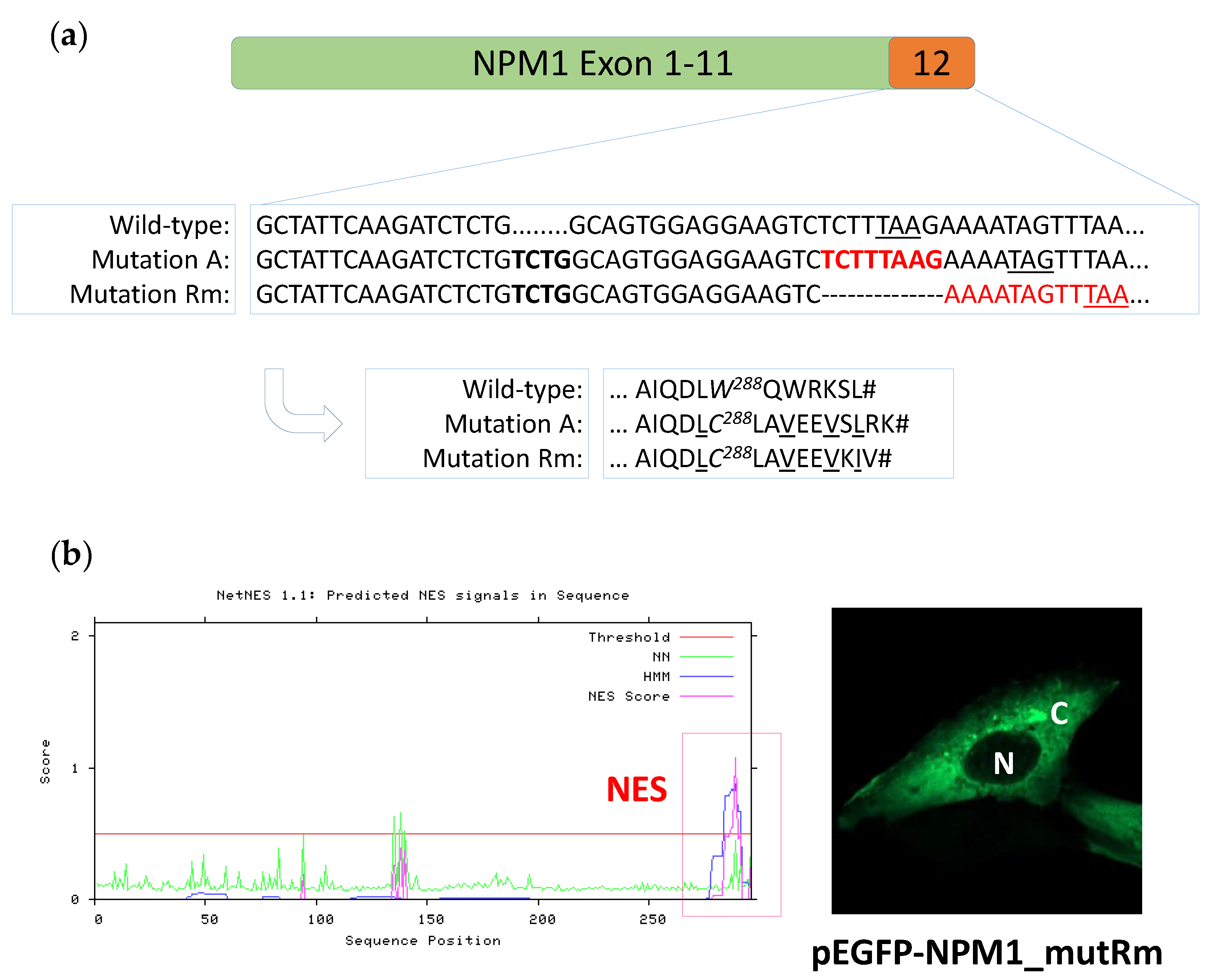
3.1. Identification of a New NPM1 Mutation Involving Exon 12
3.2. The New NPM1 Mutant Protein Displays a New C-Terminal Nuclear Export Signal (NES) Motif
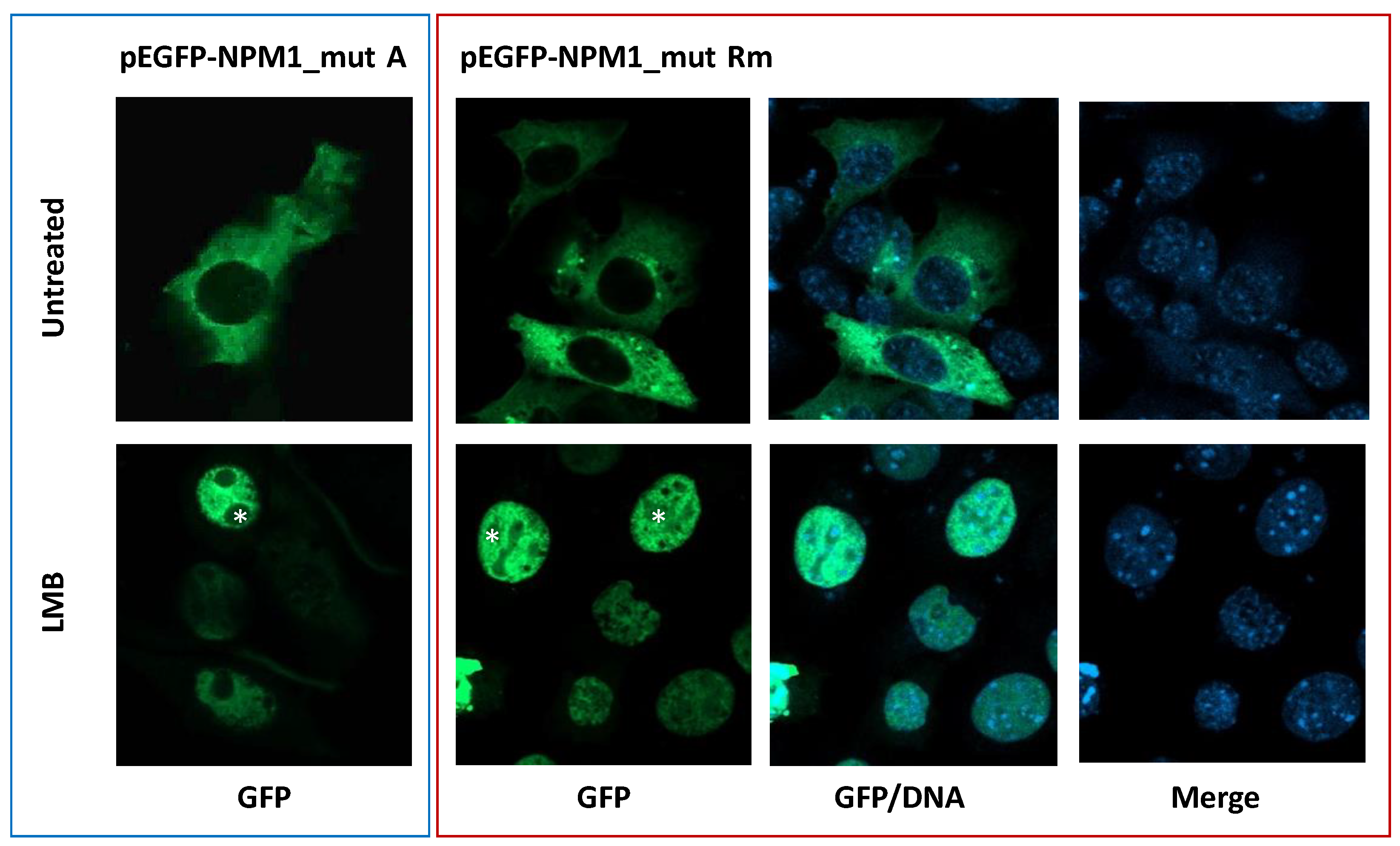
3.3. The New NPM1 Mutant Protein Locates in the Cytoplasm in a NES-Dependent Manner
3.4. Detection of NPM1 Mutant Rm Transcripts by q-RT-PCR for NPM1 Mutation A
3.5. Characteristics of AML Carrying NPM1 Mutation Rm
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Falini, B.; Mecucci, C.; Tiacci, E.; Alcalay, M.; Rosati, R.; Pasqualucci, L.; La Starza, R.; Diverio, D.; Colombo, E.; Santucci, A.; et al. Cytoplasmic nucleophosmin in acute myelogenous leukemia with a normal karyotype. N. Engl. J. Med. 2005, 352, 254–266. [Google Scholar] [CrossRef]
- Falini, B.; Nicoletti, I.; Martelli, M.F.; Mecucci, C. Acute myeloid leukemia carrying cytoplasmic/mutated nucleophosmin (NPMc+ AML): Biologic and clinical features. Blood 2007, 109, 874–885. [Google Scholar] [CrossRef] [Green Version]
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016, 127, 2391–2405. [Google Scholar] [CrossRef]
- Grisendi, S.; Mecucci, C.; Falini, B.; Pandolfi, P.P. Nucleophosmin and cancer. Nat. Rev. Cancer 2006, 6, 493–505. [Google Scholar] [CrossRef]
- Grisendi, S.; Bernardi, R.; Rossi, M.; Cheng, K.; Khandker, L.; Manova, K.; Pandolfi, P.P. Role of nucleophosmin in embryonic development and tumorigenesis. Nature 2005, 437, 147–153. [Google Scholar] [CrossRef]
- Boulon, S.; Westman, B.J.; Hutten, S.; Boisvert, F.M.; Lamond, A.I. The nucleolus under stress. Mol. Cell 2010, 40, 216–227. [Google Scholar] [CrossRef]
- Falini, B.; Martelli, M.P.; Mecucci, C.; Liso, A.; Bolli, N.; Bigerna, B.; Pucciarini, A.; Pileri, S.; Meloni, G.; Martelli, M.F.; et al. Cytoplasmic mutated nucleophosmin is stable in primary leukemic cells and in a xenotransplant model of NPMc+ acute myeloid leukemia in SCID mice. Haematologica 2008, 93, 775–779. [Google Scholar] [CrossRef] [Green Version]
- McKerrell, T.; Park, N.; Moreno, T.; Grove, C.S.; Ponstingl, H.; Stephens, J.; Crawley, C.; Craig, J.; Scott, M.A.; Hodkinson, C.; et al. Leukemia-associated somatic mutations drive distinct patterns of age-related clonal hemopoiesis. Cell Rep. 2015, 10, 1239–1245. [Google Scholar] [CrossRef]
- Falini, B.; Sportoletti, P. A scale of bad co-mutations in NPM1-driven AML. Blood 2017, 130, 1877–1879. [Google Scholar] [CrossRef]
- Falini, B.; Martelli, M.P. NPM1-mutated AML: Targeting by disassembling. Blood 2011, 118, 2936–2938. [Google Scholar] [CrossRef] [Green Version]
- Hingorani, K.; Szebeni, A.; Olson, M.O. Mapping the functional domains of nucleolar protein B23. J. Biol. Chem. 2000, 275, 24451–24457. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Budhu, A.; Forgues, M.; Wang, X.W. Temporal and spatial control of nucleophosmin by the Ran-Crm1 complex in centrosome duplication. Nat. Cell Biol. 2005, 7, 823–830. [Google Scholar] [CrossRef]
- Bolli, N.; Nicoletti, I.; De Marco, M.F.; Bigerna, B.; Pucciarini, A.; Mannucci, R.; Martelli, M.P.; Liso, A.; Mecucci, C.; Fabbiano, F.; et al. Born to be exported: COOH-terminal nuclear export signals of different strength ensure cytoplasmic accumulation of nucleophosmin leukemic mutants. Cancer Res. 2007, 67, 6230–6237. [Google Scholar] [CrossRef] [Green Version]
- Falini, B.; Albiero, E.; Bolli, N.; De Marco, M.F.; Madeo, D.; Martelli, M.; Nicoletti, I.; Rodeghiero, F. Aberrant cytoplasmic expression of C-terminal-truncated NPM leukaemic mutant is dictated by tryptophans loss and a new NES motif. Leukemia 2007, 21, 2052–2054; author reply 2054; discussion 2055–2056. [Google Scholar] [CrossRef] [Green Version]
- Mariano, A.R.; Colombo, E.; Luzi, L.; Martinelli, P.; Volorio, S.; Bernard, L.; Meani, N.; Bergomas, R.; Alcalay, M.; Pelicci, P.G. Cytoplasmic localization of NPM in myeloid leukemias is dictated by gain-of-function mutations that create a functional nuclear export signal. Oncogene 2006, 25, 4376–4380. [Google Scholar] [CrossRef] [Green Version]
- Albiero, E.; Madeo, D.; Bolli, N.; Giaretta, I.; Bona, E.D.; Martelli, M.F.; Nicoletti, I.; Rodeghiero, F.; Falini, B. Identification and functional characterization of a cytoplasmic nucleophosmin leukaemic mutant generated by a novel exon-11 NPM1 mutation. Leukemia 2007, 21, 1099–1103. [Google Scholar] [CrossRef] [Green Version]
- Martelli, M.P.; Rossi, R.; Venanzi, A.; Meggendorfer, M.; Perriello, V.M.; Martino, G.; Spinelli, O.; Ciurnelli, R.; Varasano, E.; Brunetti, L.; et al. Novel Npm1 Exon 5 Mutations and Gene Fusions Leading to Aberrant Cytoplasmic Nucleophosmin in Aml. Blood 2021, in press. [Google Scholar] [CrossRef]
- Brunetti, L.; Gundry, M.C.; Sorcini, D.; Guzman, A.G.; Huang, Y.H.; Ramabadran, R.; Gionfriddo, I.; Mezzasoma, F.; Milano, F.; Nabet, B.; et al. Mutant NPM1 Maintains the Leukemic State through HOX Expression. Cancer Cell 2018, 34, 499–512. [Google Scholar] [CrossRef] [Green Version]
- Falini, B.; Martelli, M.P.; Bolli, N.; Bonasso, R.; Ghia, E.; Pallotta, M.T.; Diverio, D.; Nicoletti, I.; Pacini, R.; Tabarrini, A.; et al. Immunohistochemistry predicts nucleophosmin (NPM) mutations in acute myeloid leukemia. Blood 2006, 108, 1999–2005. [Google Scholar] [CrossRef]
- Martelli, M.P.; Manes, N.; Liso, A.; Pettirossi, V.; Verducci Galletti, B.; Bigerna, B.; Pucciarini, A.; De Marco, M.F.; Pallotta, M.T.; Bolli, N.; et al. A western blot assay for detecting mutant nucleophosmin (NPM1) proteins in acute myeloid leukaemia. Leukemia 2008, 22, 2285–2288. [Google Scholar] [CrossRef] [Green Version]
- Falini, B.; Brunetti, L.; Martelli, M.P. How I diagnose and treat NPM1-mutated AML. Blood 2021, 137, 589–599. [Google Scholar] [CrossRef]
- Quentmeier, H.; Martelli, M.P.; Dirks, W.G.; Bolli, N.; Liso, A.; Macleod, R.A.; Nicoletti, I.; Mannucci, R.; Pucciarini, A.; Bigerna, B.; et al. Cell line OCI/AML3 bears exon-12 NPM gene mutation-A and cytoplasmic expression of nucleophosmin. Leukemia 2005, 19, 1760–1767. [Google Scholar] [CrossRef] [Green Version]
- Tiacci, E.; Spanhol-Rosseto, A.; Martelli, M.P.; Pasqualucci, L.; Quentmeier, H.; Grossmann, V.; Drexler, H.G.; Falini, B. The NPM1 wild-type OCI-AML2 and the NPM1-mutated OCI-AML3 cell lines carry DNMT3A mutations. Leukemia 2012, 26, 554–557. [Google Scholar] [CrossRef]
- Xu, C.; Gu, X.; Padmanabhan, R.; Wu, Z.; Peng, Q.; DiCarlo, J.; Wang, Y. smCounter2: An accurate low-frequency variant caller for targeted sequencing data with unique molecular identifiers. Bioinformatics 2019, 35, 1299–1309. [Google Scholar] [CrossRef] [Green Version]
- Falini, B.; Bolli, N.; Shan, J.; Martelli, M.P.; Liso, A.; Pucciarini, A.; Bigerna, B.; Pasqualucci, L.; Mannucci, R.; Rosati, R.; et al. Both carboxy-terminus NES motif and mutated tryptophan(s) are crucial for aberrant nuclear export of nucleophosmin leukemic mutants in NPMc+ AML. Blood 2006, 107, 4514–4523. [Google Scholar] [CrossRef] [PubMed]
- Pasqualucci, L.; Liso, A.; Martelli, M.P.; Bolli, N.; Pacini, R.; Tabarrini, A.; Carini, M.; Bigerna, B.; Pucciarini, A.; Mannucci, R.; et al. Mutated nucleophosmin detects clonal multilineage involvement in acute myeloid leukemia: Impact on WHO classification. Blood 2006, 108, 4146–4155. [Google Scholar] [CrossRef] [PubMed]
- Falini, B.; Macijewski, K.; Weiss, T.; Bacher, U.; Schnittger, S.; Kern, W.; Kohlmann, A.; Klein, H.U.; Vignetti, M.; Piciocchi, A.; et al. Multilineage dysplasia has no impact on biologic, clinicopathologic, and prognostic features of AML with mutated nucleophosmin (NPM1). Blood 2010, 115, 3776–3786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sportoletti, P.; Varasano, E.; Rossi, R.; Bereshchenko, O.; Cecchini, D.; Gionfriddo, I.; Bolli, N.; Tiacci, E.; Intermesoli, T.; Zanghi, P.; et al. The human NPM1 mutation A perturbs megakaryopoiesis in a conditional mouse model. Blood 2013, 121, 3447–3458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N. Engl. J. Med. 2013, 368, 2059–2074. [CrossRef] [Green Version]
- Eisfeld, A.K.; Kohlschmidt, J.; Mrozek, K.; Blachly, J.S.; Walker, C.J.; Nicolet, D.; Orwick, S.; Maharry, S.E.; Carroll, A.J.; Stone, R.M.; et al. Mutation patterns identify adult patients with de novo acute myeloid leukemia aged 60 years or older who respond favorably to standard chemotherapy: An analysis of Alliance studies. Leukemia 2018, 32, 1338–1348. [Google Scholar] [CrossRef]
- Borrow, J.; Dyer, S.A.; Akiki, S.; Griffiths, M.J. Molecular roulette: Nucleophosmin mutations in AML are orchestrated through N-nucleotide addition by TdT. Blood 2019, 134, 2291–2303. [Google Scholar] [CrossRef]
- Falini, B.; Martelli, M.P.; Pileri, S.A.; Mecucci, C. Molecular and alternative methods for diagnosis of acute myeloid leukemia with mutated NPM1: Flexibility may help. Haematologica 2010, 95, 529–534. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristics | Pt. Rm |
|---|---|
| Gender | Male |
| Age | 77 |
| PLT/mmc | 36,000 |
| WBC/mmc | 102,000 |
| Diagnosis | AML de novo |
| Blasts % (PB) | 40 * |
| Karyotype | NK |
| IHC | NPMc+ |
| WB (anti-NPM1mutA) | negative |
| NPM1 mutant transcripts copy n. (MRD assay) | 506 copies/100 Abl |
| Dysplastic features | yes (MK) |
| Blasts CD34 expression | negative |
| FLT3 | wild-type |
| DNMT3A | wild-type |
| IDH1/2 | wild-type |
| TET2 | p.Glu796ArgfsTer15 |
| SRSF2 | p.Pro95Arg |
| ASXL1 | p.Ser1166LysfsTer11 |
| Treatment | Decitabine |
| Outcome | disease progression/death |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Venanzi, A.; Rossi, R.; Martino, G.; Annibali, O.; Avvisati, G.; Mameli, M.G.; Sportoletti, P.; Tiacci, E.; Falini, B.; Martelli, M.P. A Curious Novel Combination of Nucleophosmin (NPM1) Gene Mutations Leading to Aberrant Cytoplasmic Dislocation of NPM1 in Acute Myeloid Leukemia (AML). Genes 2021, 12, 1426. https://doi.org/10.3390/genes12091426
Venanzi A, Rossi R, Martino G, Annibali O, Avvisati G, Mameli MG, Sportoletti P, Tiacci E, Falini B, Martelli MP. A Curious Novel Combination of Nucleophosmin (NPM1) Gene Mutations Leading to Aberrant Cytoplasmic Dislocation of NPM1 in Acute Myeloid Leukemia (AML). Genes. 2021; 12(9):1426. https://doi.org/10.3390/genes12091426
Chicago/Turabian StyleVenanzi, Alessandra, Roberta Rossi, Giovanni Martino, Ombretta Annibali, Giuseppe Avvisati, Maria Grazia Mameli, Paolo Sportoletti, Enrico Tiacci, Brunangelo Falini, and Maria Paola Martelli. 2021. "A Curious Novel Combination of Nucleophosmin (NPM1) Gene Mutations Leading to Aberrant Cytoplasmic Dislocation of NPM1 in Acute Myeloid Leukemia (AML)" Genes 12, no. 9: 1426. https://doi.org/10.3390/genes12091426