
Using Functional Annotations to Study Pairwise Interactions in Urinary Tract Infection Communities

 , and
, and
Abstract
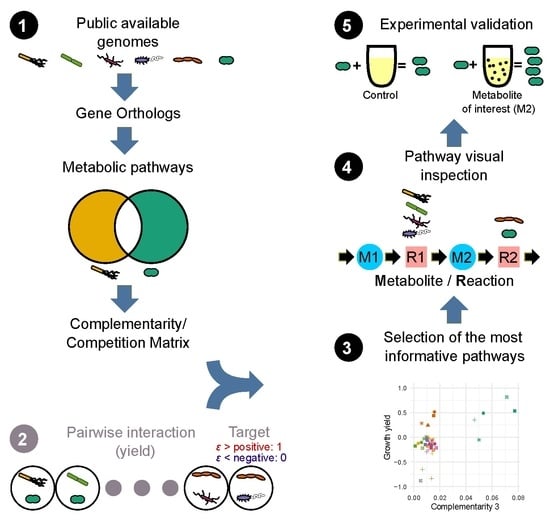
:
1. Introduction
2. Materials and Methods
2.1. Yield and Growth Rate in UTI
2.2. Description of the Dataset
2.3. Functional Annotation of the Genomes
2.4. Functional Annotation Division into Smaller Segments
2.5. Interaction Matrices
2.5.1. Complementarity
- Complementarity 1: directly as the fraction of the KOs that are present in the donor set but not in the acceptor;
- Complementarity 2: as the fraction of the total amount of KOs that the acceptor could have when complemented (by the donor) divided by the acceptor’s own possibilities;
- Complementarity 3: as Complementarity 2, but without taking into consideration the shared KOs (similar to Hamming distance for boolean vectors, but divided by A instead of total number of dimensions to make it an asymmetric index);
- Complementarity 4: same as Complementary 3, but divided by the putative total functionality. In this way, it is equivalent to Hamming distance, but it is symmetric.
2.5.2. Competition
- Competition 1 : directly the fraction of shared KOs;
- Competition 2: equivalent to competition_1 but compared to the acceptor function to make it asymmetric;
- Competition 3: equivalent to competition_2, but compared to the total possibilities;
- Competition 4: is just Pearson correlation (just for comparison).
2.5.3. Selection of Interaction Indices
2.5.4. Selection of Informative Pathways
2.6. Alternative Approaches
2.7. Checking Transporters Availability
2.8. Supplementation Experiments
3. Results
3.1. Complementarity Indices Show the Greatest Correlation with the Pairwise Interaction Growth Yields
3.2. Analysis of Genomes in Terms of KEGG Pathways
3.3. Enterococci Are Metabolically Complemented by the UTI Community
3.4. Folic Acid Promotes Growth of Enterococci in Artificial Urine Medium
3.5. Alternative Approaches Fail to Discover Relevant Patterns
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Frank, D.N.; Zhu, W.; Sartor, R.B.; Li, E. Investigating the biological and clinical significance of human dysbioses. Trends Microbiol. 2011, 19, 427–434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faust, K.; Raes, J. Microbial interactions: From networks to models. Nat. Rev. Microbiol. 2012, 10, 538–550. [Google Scholar] [CrossRef] [PubMed]
- Pande, S.; Kaftan, F.; Lang, S.; Svatoš, A.; Germerodt, S.; Kost, C. Privatization of cooperative benefits stabilizes mutualistic cross-feeding interactions in spatially structured environments. ISME J. 2015, 10, 1413–1423. [Google Scholar] [CrossRef] [PubMed]
- D’Souza, G.; Shitut, S.; Preussger, D.; Yousif, G.; Waschina, S.; Kost, C. Ecology and evolution of metabolic cross-feeding interactions in bacteria. Nat. Prod. Rep. 2018, 35, 455–488. [Google Scholar] [CrossRef] [Green Version]
- De Vos, M.G.J.; Zagorski, M.; McNally, A.; Bollenbach, T. Interaction networks, ecological stability, and collective antibiotic tolerance in polymicrobial infections. Proc. Natl. Acad. Sci. USA 2017, 114, 10666–10671. [Google Scholar] [CrossRef] [Green Version]
- Zengler, K.; Palsson, B.O. A road map for the development of community systems (CoSy) biology. Nat. Rev. Microbiol. 2012, 10, 366–372. [Google Scholar] [CrossRef]
- Perez-Garcia, O.; Lear, G.; Singhal, N. Metabolic network modeling of microbial interactions in natural and engineered environmental systems. Front. Microbiol. 2016, 7, 673. [Google Scholar] [CrossRef]
- Gottstein, W.; Olivier, B.G.; Bruggeman, F.J.; Teusink, B. Constraint-based stoichiometric modelling from single organisms to microbial communities. J. R. Soc. Interface 2016, 13, 20160627. [Google Scholar] [CrossRef]
- Hanemaaijer, M.; Roling, W.F.M.; Olivier, B.G.; Khandelwal, R.A.; Teusink, B.; Bruggeman, F.J. Systems modeling approaches for microbial community studies: From metagenomics to inference of the community structure. Front. Microbiol. 2015, 19, 213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernstein, D.B.; Sulheim, S.; Almaas, E.; Segrè, D. Addressing uncertainty in genome-scale metabolic model reconstruction and analysis. Genome Biol. 2021, 22, 64. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S.; Sato, Y.; Furumichi, M.; Tanabe, M. KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res. 2012, 40, D109–D114. [Google Scholar] [CrossRef] [Green Version]
- Caspi, R.; Altman, T.; Billington, R.; Dreher, K.; Foerster, H.; Fulcher, C.A.; Holland, T.A.; Keseler, I.M.; Kothari, A.; Kubo, A.; et al. The MetaCyc database of metabolic pathways and enzymes and the BioCyc collection of Pathway/Genome Databases. Nucleic Acids Res. 2014, 42, 459–471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Overbeek, R.; Olson, R.; Pusch, G.D.; Olsen, G.J.; Davis, J.J.; Disz, T.; Edwards, R.A.; Gerdes, S.; Parrello, B.; Shukla, M.; et al. The SEED and the Rapid Annotation of microbial genomes using Subsystems Technology (RAST). Nucleic Acids Res. 2014, 42, 206–214. [Google Scholar] [CrossRef] [PubMed]
- Huerta-Cepas, J.; Szklarczyk, D.; Forslund, K.; Cook, H.; Heller, D.; Walter, M.C.; Rattei, T.; Mende, D.R.; Sunagawa, S.; Kuhn, M.; et al. eggNOG 4.5: A hierarchical orthology framework with improved functional annotations for eukaryotic, prokaryotic and viral sequences. Nucleic Acids Res. 2016, 44, D286–D293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borenstein, E.; Kupiec, M.; Feldman, M.W.; Ruppin, E. Large-scale reconstruction and phylogenetic analysis of metabolic environments. Proc. Natl. Acad. Sci. USA 2008, 105, 14482–14487. [Google Scholar] [CrossRef] [Green Version]
- Levy, R.; Carr, R.; Kreimer, A.; Freilich, S.; Borenstein, E. NetCooperate: A network-based tool for inferring host-microbe and microbe-microbe cooperation. BMC Bioinform. 2015, 16, 164. [Google Scholar] [CrossRef] [Green Version]
- Cao, Y.; Wang, Y.; Zheng, X.; Li, F.; Bo, X. RevEcoR: An R package for the reverse ecology analysis of microbiomes. BMC Bioinform. 2016, 17, 294. [Google Scholar] [CrossRef] [Green Version]
- Melkonian, C.; Gottstein, W.; Blasche, S.; Kim, Y.; Abel-Kistrup, M.; Swiegers, H.; Saerens, S.; Edwards, N.; Patil, K.; Teusink, B.; Molenaar, D. Finding Functional Differences Between Species in a Microbial Community: Case Studies in Wine Fermentation and Kefir Culture. Front Microbiol 2019, 10, 1347. [Google Scholar] [CrossRef] [Green Version]
- Croxall, G.; Weston, V.; Joseph, S.; Manning, G.; Cheetham, P.; McNally, A. Increased human pathogenic potential of Escherichia coli from polymicrobial urinary tract infections in comparison to isolates from monomicrobial culture samples. J. Med. Microbiol. 2011, 60, 102–109. [Google Scholar] [CrossRef]
- Hilt, E.E.; McKinley, K.; Pearce, M.M.; Rosenfeld, A.B.; Zilliox, M.J.; Mueller, E.R.; Brubaker, L.; Gai, X.; Wolfe, A.J.; Schreckenberger, P.C. Urine Is Not Sterile: Use of Enhanced Urine Culture Techniques To Detect Resident Bacterial Flora in the Adult Female Bladder. J. Clin. Microbiol. 2013, 52, 871–876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolfe, A.J.; Toh, E.; Shibata, N.; Rong, R.; Kenton, K.; FitzGerald, M.; Mueller, E.R.; Schreckenberger, P.; Dong, Q.; Nelson, D.E.; et al. Evidence of Uncultivated Bacteria in the Adult Female Bladder. J. Clin. Microbiol. 2012, 50, 1376–1383. [Google Scholar] [CrossRef] [Green Version]
- Khasriya, R.; Sathiananthamoorthy, S.; Ismail, S.; Kelsey, M.; Wilson, M.; Rohn, J.L.; Malone-Lee, J. Spectrum of Bacterial Colonization Associated with Urothelial Cells from Patients with Chronic Lower Urinary Tract Symptoms. J. Clin. Microbiol. 2013, 51, 2054–2062. [Google Scholar] [CrossRef] [Green Version]
- Fouts, D.E.; Pieper, R.; Szpakowski, S.; Pohl, H.; Knoblach, S.; Suh, M.J.; Huang, S.T.; Ljungberg, I.; Sprague, B.M.; Lucas, S.K.; et al. Integrated next-generation sequencing of 16S rDNA and metaproteomics differentiate the healthy urine microbiome from asymptomatic bacteriuria in neuropathic bladder associated with spinal cord injury. J. Transl. Med. 2012, 10, 174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neugent, M.L.; Hulyalkar, N.V.; Nguyen, V.H.; Zimmern, P.E.; Nisco, N.J.D. Advances in Understanding the Human Urinary Microbiome and Its Potential Role in Urinary Tract Infection. mBio 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Janes, V.A.; Matamoros, S.; Willemse, N.; Visser, C.E.; de Wever, B.; Jakobs, M.E.; Subramanian, P.; Hasan, N.A.; Colwell, R.R.; de Jong, M.D.; et al. Metagenomic sequencing to replace semi-quantitative urine culture for detection of urinary tract infections: A proof of concept. bioRxiv 2017, 178178. [Google Scholar] [CrossRef]
- Stamm, W.E.; Norrby, S.R. Urinary tract infections: Disease panorama and challenges. J. Infect. Dis. 2001, 183, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Foxman, B. Epidemiology of urinary tract infections: Incidence, morbidity, and economic costs. Am. J. Med. 2002, 113, 5–13. [Google Scholar] [CrossRef]
- Medina, M.; Castillo-Pino, E. An introduction to the epidemiology and burden of urinary tract infections. Ther. Adv. Urol. 2019, 11, 175628721983217. [Google Scholar] [CrossRef] [Green Version]
- Flores-Mireles, A.L.; Walker, J.N.; Caparon, M.; Hultgren, S.J. Urinary tract infections: Epidemiology, mechanisms of infection and treatment options. Nat. Rev. Microbiol. 2015, 13, 269–284. [Google Scholar] [CrossRef] [PubMed]
- Imirzalioglu, C.; Hain, T.; Chakraborty, T.; Domann, E. Hidden pathogens uncovered: Metagenomic analysis of urinary tract infections. Andrologia 2008, 40, 66–71. [Google Scholar] [CrossRef] [PubMed]
- Zandbergen, L.E.; Halverson, T.; Brons, J.K.; Wolfe, A.J.; de Vos, M.G.J. The Good and the Bad: Ecological Interaction Measurements Between the Urinary Microbiota and Uropathogens. Front. Microbiol. 2021, 12, 1026. [Google Scholar] [CrossRef] [PubMed]
- Brooks, T.; Keevil, C.W. A simple artificial urine for the growth of urinary pathogens. Lett. Appl. Microbiol. 1997, 24, 203–206. [Google Scholar] [CrossRef] [PubMed]
- NCBI Resource Coordinators. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2016, 44, D7–D19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKinney, W. Pandas: A Foundational Python Library for Data Analysis and Statistics. Python High Perform. Sci. Comput. 2011, 14, 1–9. [Google Scholar]
- DeSantis, T.Z.; Hugenholtz, P.; Larsen, N.; Rojas, M.; Brodie, E.L.; Keller, K.; Huber, T.; Dalevi, D.; Hu, P.; Andersen, G.L. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl. Environ. Microbiol. 2006, 72, 5069–5072. [Google Scholar] [CrossRef] [Green Version]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Benson, D.A.; Karsch-Mizrachi, I.; Lipman, D.J.; Ostell, J.; Wheeler, D.L. GenBank. Nucleic Acids Res. 2005, 33, D34–D38. [Google Scholar] [CrossRef] [Green Version]
- Hyatt, D.; Chen, G.L.; LoCascio, P.F.; Land, M.L.; Larimer, F.W.; Hauser, L.J. Prodigal: Prokaryotic gene recognition and translation initiation site identification. BMC Bioinform. 2010, 11, 119. [Google Scholar] [CrossRef] [Green Version]
- Kanehisa, M.; Sato, Y.; Morishima, K. BlastKOALA and GhostKOALA: KEGG Tools for Functional Characterization of Genome and Metagenome Sequences. J. Mol. Biol. 2016, 428, 726–731. [Google Scholar] [CrossRef] [Green Version]
- Markowitz, V.M.; Chen, I.M.A.; Palaniappan, K.; Chu, K.; Szeto, E.; Grechkin, Y.; Ratner, A.; Jacob, B.; Huang, J.; Williams, P.; et al. IMG: The Integrated Microbial Genomes database and comparative analysis system. Nucleic Acids Res. 2012, 40, D115–D122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.; Ma, Y. Ensemble Machine Learning: Methods and Applications; Springer: New York, NY, USA, 2012; p. 329. [Google Scholar]
- Guyon, I.; Weston, J.; Barnhill, S.; Vapnik, V. Gene Selection for Cancer Classification using Support Vector Machines. Mach. Learn. 2002, 46, 389–422. [Google Scholar] [CrossRef]
- Kursa, M.B.; Rudnicki, W.R. Feature Selection with the Boruta Package. J. Stat. Softw. 2010, 36. [Google Scholar] [CrossRef] [Green Version]
- Kanehisa, M.; Sato, Y.; Kawashima, M.; Furumichi, M.; Tanabe, M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 2015, 44, D457–D462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Portela, C.A.F.; Smart, K.F.; Tumanov, S.; Cook, G.M.; Villas-Bôas, S.G. Global metabolic response of Enterococcus faecalis to oxygen. J. Bacteriol. 2014, 196, 2012–2022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zervos, M.J.; Schaberg, D.R. Trimethoprim-Sulfamethoxazole by Folinic Acid. Antimicrob. Agents Chemother. 1985, 28, 446–448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chenoweth, C.E.; Robinson, K.A.; Schaberg, D.R. Efficacy of Ampicillin versus Trimethoprim-Sulfamethoxazole in a Mouse Model of Lethal Enterococcal Peritonitis. Antimicrob. Agents Chemother. 1990, 34, 1800–1802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burke, C.; Steinberg, P.; Rusch, D.; Kjelleberg, S.; Thomas, T. Bacterial community assembly based on functional genes rather than species. Proc. Natl. Acad. Sci. USA 2011, 108, 14288–14293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Konstantinidis, K.T.; Tiedje, J.M. Trends between gene content and genome size in prokaryotic species with larger genomes. Proc. Natl. Acad. Sci. USA 2003, 101, 3160–3165. [Google Scholar] [CrossRef] [Green Version]
- Cardona, C.; Weisenhorn, P.; Henry, C.; Gilbert, J.A. Network-based metabolic analysis and microbial community modeling. Curr. Opin. Microbiol. 2016, 31, 124–131. [Google Scholar] [CrossRef]
- Layeghifard, M.; Hwang, D.M.; Guttman, D.S. Disentangling Interactions in the Microbiome: A Network Perspective. Trends Microbiol. 2017, 25, 217–228. [Google Scholar] [CrossRef]
- Machado, D.; Maistrenko, O.M.; Andrejev, S.; Kim, Y.; Bork, P.; Patil, K.R.; Patil, K.R. Polarization of microbial communities between competitive and cooperative metabolism. Nat. Ecol. Evol. 2021, 5, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Kuczynski, J.; Liu, Z.; Lozupone, C.; McDonald, D.; Fierer, N.; Knight, R. Microbial community resemblance methods differ in their ability to detect biologically relevant patterns. Nat. Methods 2010, 7, 813–819. [Google Scholar] [CrossRef] [PubMed]
- Foster, K.R.; Bell, T. Competition, not cooperation, dominates interactions among culturable microbial species. Curr. Biol. 2012, 22, 1845–1850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, Q.; Chen, K.; Paulsen, I.T. TransportDB: A comprehensive database resource for cytoplasmic membrane transport systems and outer membrane channels. Nucleic Acids Res. 2007, 35, D274–D279. [Google Scholar] [CrossRef] [PubMed]
- Veith, N.; Solheim, M.; van Grinsven, K.W.A.; Olivier, B.G.; Levering, J.; Grosseholz, R.; Hugenholtz, J.; Holo, H.; Nes, I.; Teusink, B.; Kummer, U. Using a genome-scale metabolic model of Enterococcus faecalis V583 to assess amino acid uptake and its impact on central metabolism. Appl. Environ. Microbiol. 2015, 81, 1622–1633. [Google Scholar] [CrossRef] [Green Version]
- Kurutas, E.B.; Ciragil, P.; Gul, M.; Kilinc, M. The effects of oxidative stress in urinary tract infection. Mediat. Inflamm. 2005, 2005, 242–244. [Google Scholar] [CrossRef]
- Levy, R.; Borenstein, E. Metabolic modeling of species interaction in the human microbiome elucidates community-level assembly rules. Proc. Natl. Acad. Sci. USA 2013, 110, 12804–12809. [Google Scholar] [CrossRef] [Green Version]
- Lipson, D.A. The complex relationship between microbial growth rate and yield and its implications for ecosystem processes. Front. Microbiol. 2015, 6, 615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bairey, E.; Kelsic, E.D.; Kishony, R. High-order species interactions shape ecosystem diversity. Nat. Commun. 2016, 7, 12285. [Google Scholar] [CrossRef]
- Guo, X.; Boedicker, J.Q. The contribution of high-order metabolic interactions to the global activity of a four-species microbial community. PLOS Comput. Biol. 2016, 12, e1005079. [Google Scholar] [CrossRef] [PubMed]
- Nielubowicz, G.R.; Mobley, H.L.T. Host–pathogen interactions in urinary tract infection. Nat. Rev. Urol. 2010, 7, 430–441. [Google Scholar] [CrossRef] [PubMed]
- Lima, W.C.; Balestrino, D.; Forestier, C.; Cosson, P. Two distinct sensing pathways allow recognition of K lebsiella pneumoniae by D ictyostelium amoebae. Cell. Microbiol. 2013, 16, 311–323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdelsalam, I. Production of vitamin B12 and folate using a potent mutant strain of Klebsiella pneumonia. Egypt. J. Chem. 2018, 61, 93–100. [Google Scholar] [CrossRef]
- Kariluoto, S.; Edelmann, M.; Herranen, M.; Lampi, A.M.; Shmelev, A.; Salovaara, H.; Korhola, M.; Piironen, V. Production of folate by bacteria isolated from oat bran. Int. J. Food Microbiol. 2010, 143, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Masters, P.A.; O’Bryan, T.A.; Zurlo, J.; Miller, D.Q.; Joshi, N. Trimethoprim-Sulfamethoxazole Revisited. Arch. Intern. Med. 2003, 163, 402. [Google Scholar] [CrossRef]
- Hollenbeck, B.L.; Rice, L.B. Intrinsic and acquired resistance mechanisms in enterococcus. Virulence 2012, 3, 421–569. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pathways | RF | SVM | MWU | Ensemble | ||||
|---|---|---|---|---|---|---|---|---|
| Feat. Imp. | Rank | Coef. | Rank | p-Values | Rank | Sum | Rank | |
| Lipopolysaccharide biosynthesis | 0.111 | 1 | 2.265 | 1 | 0.0003 | 13 | 15 | 1 |
| Tyrosine metabolism | 0.028 | 11 | 0.531 | 6 | 3.08 × | 1 | 18 | 2 |
| Sulfur metabolism | 0.058 | 6 | 0.888 | 3 | 0.0003 | 12 | 21 | 3 |
| Cationic antimicrobial peptide (CAMP) resistance | 0.036 | 9 | 0.513 | 9 | 0.0001 | 6 | 24 | 4 |
| Phenylalanine metabolism | 0.073 | 4 | 0.313 | 16 | 0.0002 | 10 | 30 | 5 |
| Biotin metabolism | 0.017 | 15 | 0.385 | 14 | 7.01 × | 4 | 33 | 6 |
| Glutathione metabolism | 0.015 | 17 | 0.243 | 22 | 9.44 × | 5 | 44 | 7 |
| Bacterial secretion system | 0.050 | 7 | 0.905 | 2 | 0.001 | 41 | 50 | 8 |
| Biofilm formation-Pseudomonas aeruginosa | 0.020 | 14 | 0.318 | 15 | 0.0007 | 29 | 58 | 9 |
| Riboflavin metabolism | 0.007 | 23 | 0.518 | 8 | 0.0007 | 28 | 59 | 10 |
| Histidine metabolism | 0.014 | 18 | −0.107 | 65 | 0.0003 | 14 | 97 | 22 |
| Folate biosynthesis | 0.001 | 53 | 0.016 | 48 | 0.0003 | 15 | 116 | 31 |
| Compound | Biological Relevance |
|---|---|
| Fumarate | Two ways to produce fumarate: from L-Aspartate (1) and from citruline (2), are not present in enterococci and staphylococci, but are in the others. |
| Sulphide | Sulphide can be formed from extracellular sulfate by many forms. Ent and Ecoli do not have the necessary orthologs, but they are able to convert this sulphide in Acetate and L-Cysteine (3). Cysteine is an amino acid, which has antioxidant activity [45]. |
| Histidine, Glutamate, and Serine | Not directly synthesised in Enterococcus (4, 4, 5, respectively). |
| Folate (DHF) and derivates (10 formyl THF) | Routes available for production of folate (DHF) and derivates (10formylTHF) were missing in Enterococcus but not in the other genera. (6 (folate) and 7 (derivates)). Folate is an essential cofactor for nucleotide and amino acid biosynthesis. This makes folate a candidate for the hypothesis of complementarity. In addition, in mammalian cells there are folate transporters, however, in most bacteria those transporters do not exist and they need to synthesise their own folate, which make the biosynthesis of folate a target for antibiotics. However, some strains of enterococci are able to uptake exogenous folate [46,47], which would exclude them for the antibiotic target. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lara, E.G.; van der Windt, I.; Molenaar, D.; de Vos, M.G.J.; Melkonian, C. Using Functional Annotations to Study Pairwise Interactions in Urinary Tract Infection Communities. Genes 2021, 12, 1221. https://doi.org/10.3390/genes12081221
Lara EG, van der Windt I, Molenaar D, de Vos MGJ, Melkonian C. Using Functional Annotations to Study Pairwise Interactions in Urinary Tract Infection Communities. Genes. 2021; 12(8):1221. https://doi.org/10.3390/genes12081221
Chicago/Turabian StyleLara, Elena G., Isabelle van der Windt, Douwe Molenaar, Marjon G. J. de Vos, and Chrats Melkonian. 2021. "Using Functional Annotations to Study Pairwise Interactions in Urinary Tract Infection Communities" Genes 12, no. 8: 1221. https://doi.org/10.3390/genes12081221