
Harnessing the Nucleolar DNA Damage Response in Cancer Therapy


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. rDNA Structure
2.1. rDNA: An Intrinsically Unstable Genomic Region
2.2. rDNA Instability in Cancer
3. Mechanisms of rDNA Repair
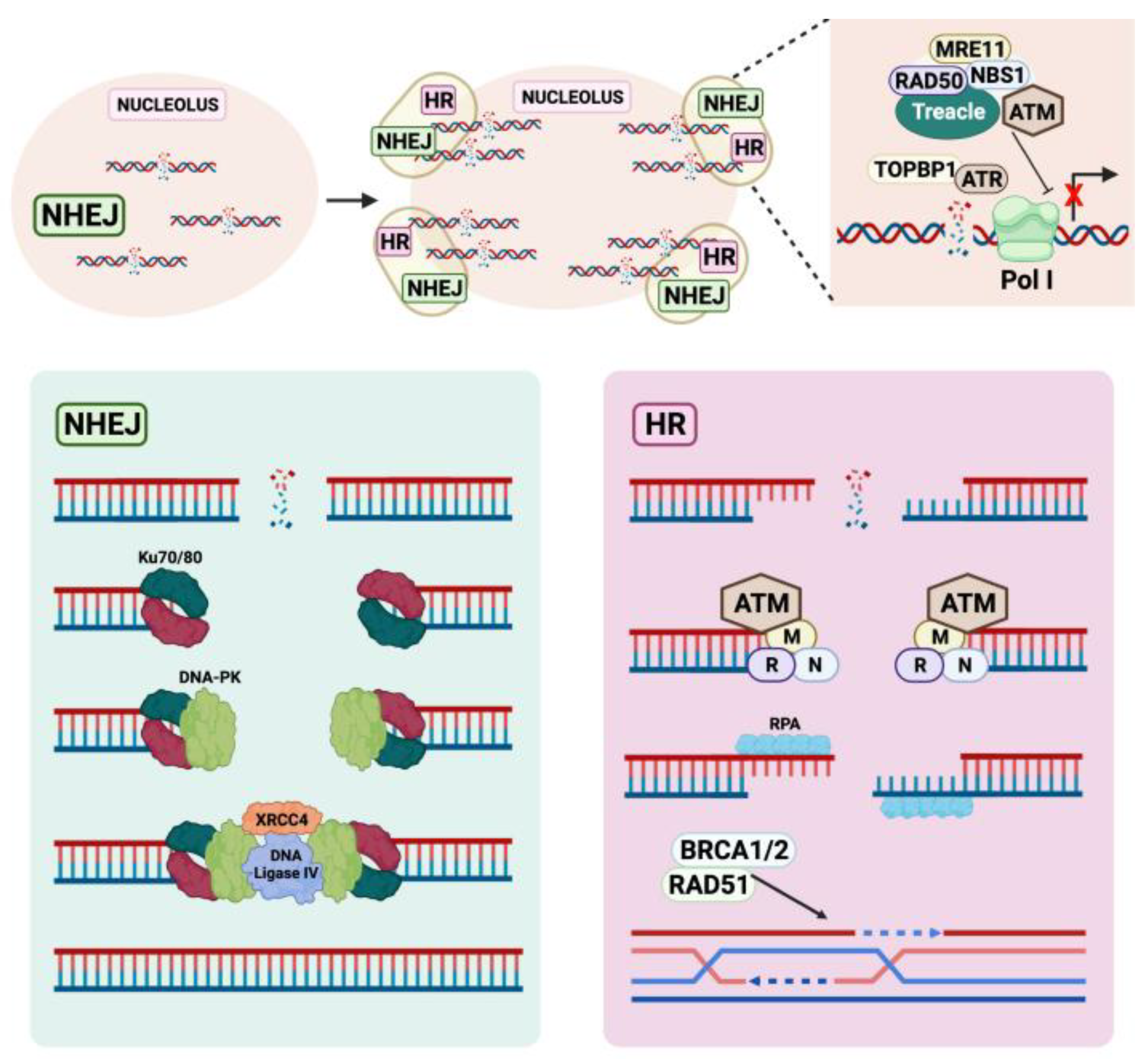
4. The Crosstalk between the Nucleolus and the DDR
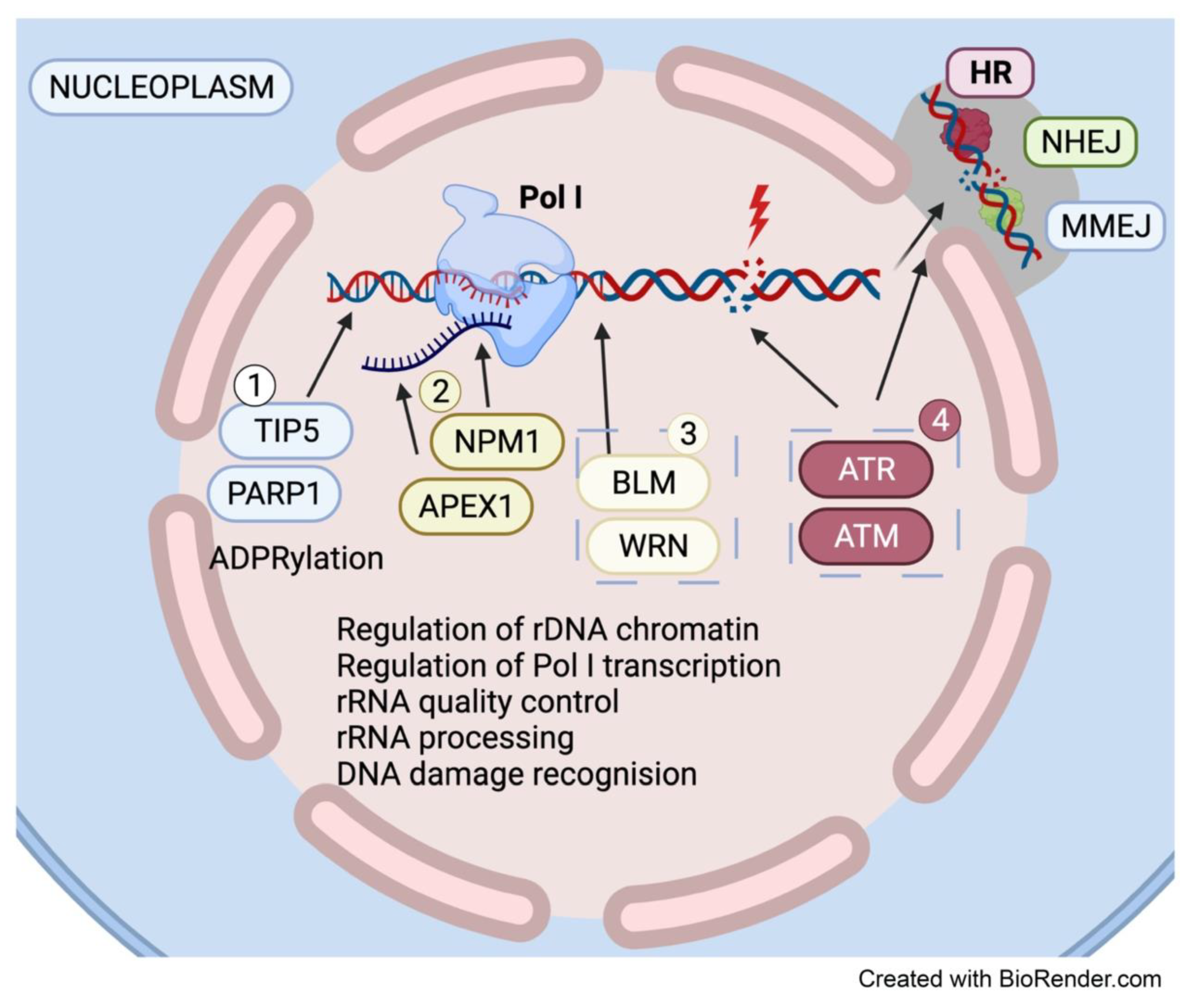
4.1. The Nucleolus Is a Hub for DNA Repair Factors
4.2. DNA Damage-Induced Nucleolar-Nucleoplasmic Shuttling
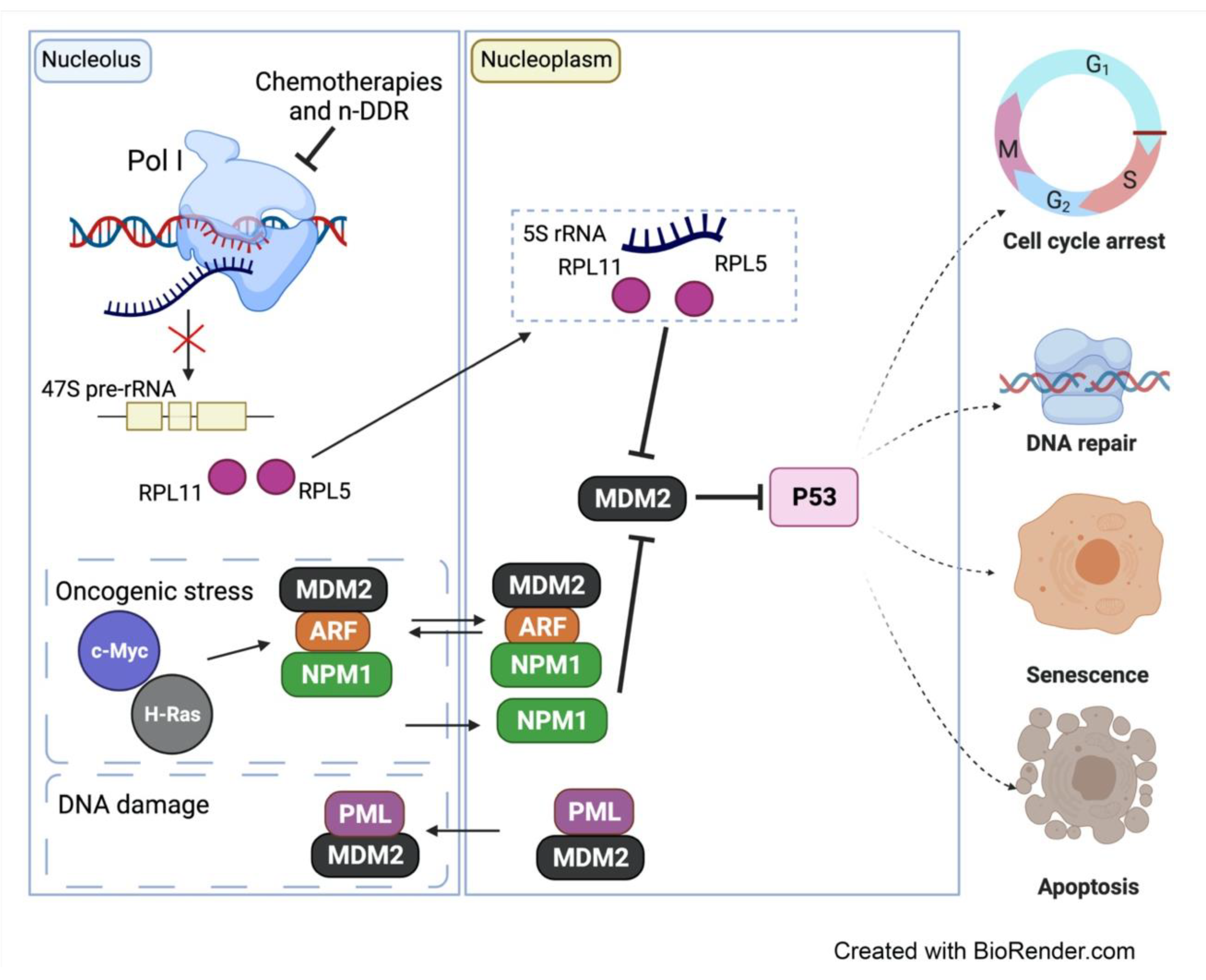
5. The Nucleolar DDR in Cancer Therapy
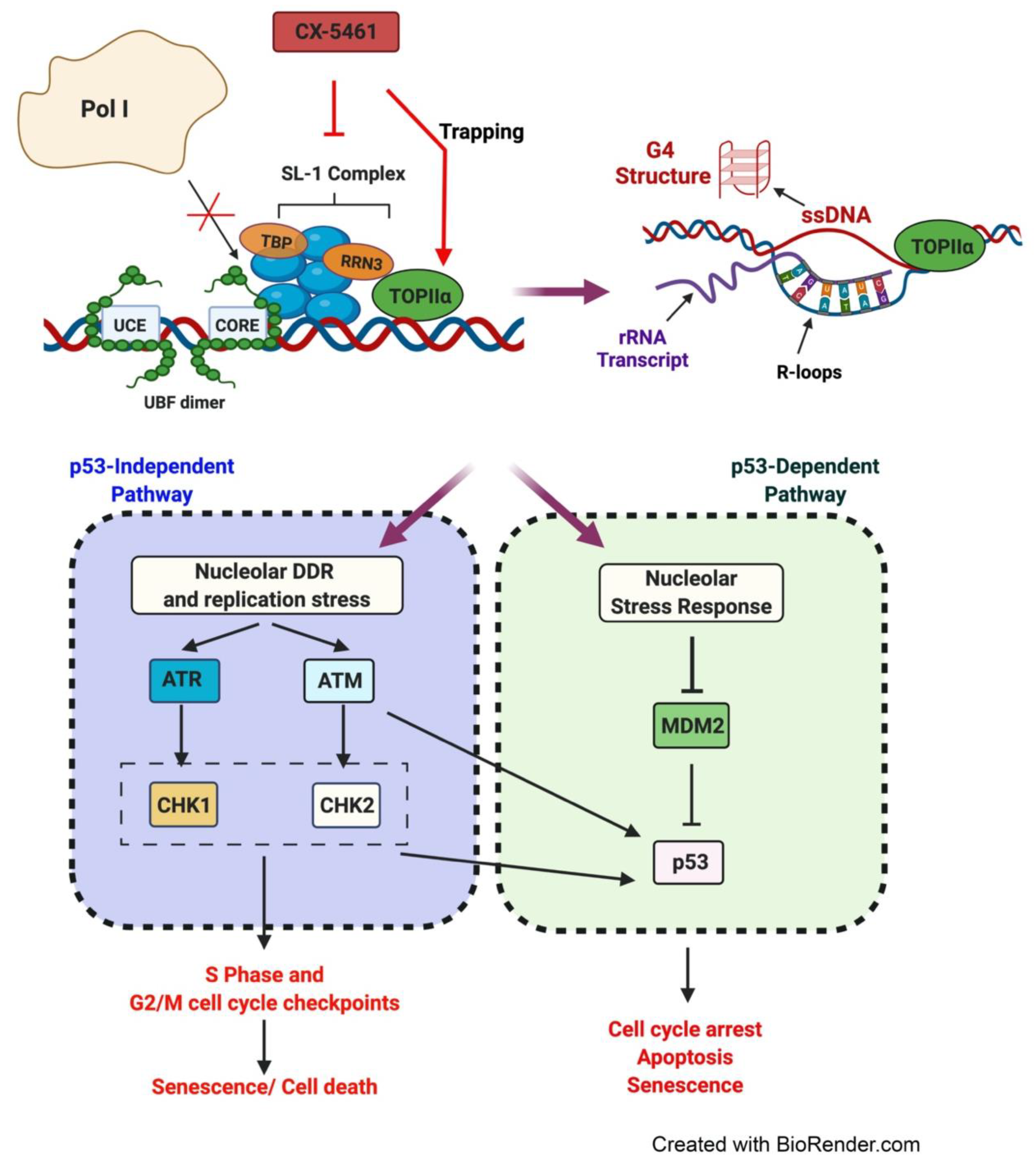
5.1. CX-5461′s Mode of Action
5.2. Therapeutic Response to CX-5461 Is Mediated via Activation of the IRBC and the n-DDR
5.3. CX-5461 in Combination with Standard of Care Cancer Therapies
6. Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Derenzini, M.; Thiry, M.; Goessens, G. Ultrastructural cytochemistry of the mammalian cell nucleolus. J. Histochem. Cytochem. 1990, 38, 1237–1256. [Google Scholar] [CrossRef]
- Puvion-Dutilleul, F.; Puvion, E.; Bachellerie, J.P. Early stages of pre-rRNA formation within the nucleolar ultrastructure of mouse cells studied by in situ hybridization with a 5’ETS leader probe. Chromosoma 1997, 105, 496–505. [Google Scholar] [CrossRef] [PubMed]
- Fatica, A.; Tollervey, D. Making ribosomes. Curr. Opin. Cell Biol. 2002, 14, 313–318. [Google Scholar] [CrossRef]
- Henras, A.; Plisson-Chastang, C.; O’Donohue, M.-F.; Chakraborty, A.; Gleizes, P.-E. An overview of pre-ribosomal RNA processing in eukaryotes. Wiley Interdiscip. Rev. RNA 2015, 6, 225–242. [Google Scholar] [CrossRef] [PubMed]
- Hein, N.; Sanij, E.; Quin, J.; Hannan, K.M.; Hannan, R.D. The Nucleolus and Ribosomal Genes in Aging and Senescence. Senecence 2012, 171–208. [Google Scholar]
- Boulon, S.; Westman, B.J.; Hutten, S.; Boisvert, F.-M.; Lamond, A.I. The Nucleolus under Stress. Mol. Cell 2010, 40, 216–227. [Google Scholar] [CrossRef]
- Ghoshal, K.; Jacob, S.T. Specific inhibition of pre-ribosomal RNA processing in extracts from the lymphosarcoma cells treated with 5-fluorouracil. Cancer Res. 1994, 54, 632–636. [Google Scholar] [PubMed]
- Treiber, D.K.; Zhai, X.; Jantzen, H.M.; Essigmann, J.M. Cisplatin-DNA adducts are molecular decoys for the ribosomal RNA transcription factor hUBF (human upstream binding factor). Proc. Natl. Acad. Sci. USA 1994, 91, 5672–5676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trask, D.K.; Muller, M.T. Stabilization of type I topoisomerase-DNA covalent complexes by actinomycin D. Proc. Natl. Acad. Sci. USA 1988, 85, 1417–1421. [Google Scholar] [CrossRef] [Green Version]
- Burger, K.; Mühl, B.; Harasim, T.; Rohrmoser, M.; Malamoussi, A.; Orban, M.; Kellner, M.; Gruber-Eber, A.; Kremmer, E.; Hölzel, M.; et al. Chemotherapeutic Drugs Inhibit Ribosome Biogenesis at Various Levels. J. Biol. Chem. 2010, 285, 12416–12425. [Google Scholar] [CrossRef] [Green Version]
- Catez, F.; Venezia, N.D.; Marcel, V.; Zorbas, C.; Lafontaine, D.L.J.; Diaz, J.-J. Ribosome biogenesis: An emerging druggable pathway for cancer therapeutics. Biochem. Pharmacol. 2019, 159, 74–81. [Google Scholar] [CrossRef] [Green Version]
- Grob, A.; Colleran, C.; McStay, B. Construction of synthetic nucleoli in human cells reveals how a major functional nuclear domain is formed and propagated through cell division. Genes Dev. 2014, 28, 220–230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McStay, B.; Grummt, I. The Epigenetics of rRNA Genes: From Molecular to Chromosome Biology. Annu. Rev. Cell Dev. Biol. 2008, 24, 131–157. [Google Scholar] [CrossRef] [Green Version]
- Prokopowich, C.D.; Gregory, T.R.; Crease, T. The correlation between rDNA copy number and genome size in eukaryotes. Genome 2003, 46, 48–50. [Google Scholar] [CrossRef] [PubMed]
- Sakai, K.; Ohta, T.; Minoshima, S.; Kudoh, J.; Wang, Y.; de Jong, P.J.; Shimizu, N. Human ribosomal RNA gene cluster: Iden-tification of the proximal end containing a novel tandem repeat sequence. Genomics 1995, 26, 521–526. [Google Scholar] [CrossRef]
- Floutsakou, I.; Agrawal, S.; Nguyen, T.; Seoighe, C.; Ganley, A.; McStay, B. The shared genomic architecture of human nucleolar organizer regions. Genome Res. 2013, 23, 2003–2012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamdane, N.; Stefanovsky, V.Y.; Tremblay, M.G.; Nemeth, A.; Paquet, E.; Lessard, F.; Sanij, E.; Hannan, R.; Moss, T. Condi-tional inactivation of Upstream Binding Factor reveals its epigenetic functions and the existence of a somatic nucleolar pre-cursor body. PLOS Genet. 2014, 10, e1004505. [Google Scholar] [CrossRef] [Green Version]
- Hamperl, S.; Wittner, M.; Babl, V.; Perez-Fernandez, J.; Tschochner, H.; Griesenbeck, J. Chromatin states at ribosomal DNA loci. Biochim Biophys Acta 2013, 1829, 405–417. [Google Scholar] [CrossRef]
- Sanij, E.; Hannan, R.D. The role of UBF in regulating the structure and dynamics of transcriptionally active rDNA chroma-tin. Epigenetics 2009, 4, 374–382. [Google Scholar] [CrossRef] [PubMed]
- Sanij, E.; Poortinga, G.; Sharkey, K.; Hung, S.; Holloway, T.P.; Quin, J.; Robb, E.; Wong, L.H.; Thomas, W.; Stefanovsky, V.; et al. UBF levels determine the number of active ribosomal RNA genes in mammals. J. Cell Biol. 2008, 183, 1259–1274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moss, T.; Stefanovsky, V.Y. At the Center of Eukaryotic Life. Cell 2002, 109, 545–548. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, T. Regulation of ribosomal RNA gene copy number and its role in modulating genome integrity and evolutionary adaptability in yeast. Cell. Mol. Life Sci. 2011, 68, 1395–1403. [Google Scholar] [CrossRef] [Green Version]
- Salim, D.; Gerton, J.L. Ribosomal DNA instability and genome adaptability. Chromosom. Res. 2019, 27, 73–87. [Google Scholar] [CrossRef]
- Stults, D.M.; Killen, M.W.; Pierce, H.H.; Pierce, A.J. Genomic architecture and inheritance of human ribosomal RNA gene clusters. Genome Res. 2007, 18, 13–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Diesch, J.; Hannan, R.D.; Sanij, E. Perturbations at the ribosomal genes loci are at the centre of cellular dysfunction and hu-man disease. Cell Biosci 2014, 4, 43. [Google Scholar] [CrossRef] [Green Version]
- Xu, B.; Li, H.; Perry, J.M.; Singh, V.P.; Unruh, J.; Yu, Z.; Zakari, M.; McDowell, W.; Li, L.; Gerton, J.L. Ribosomal DNA copy number loss and sequence variation in cancer. PLOS Genet. 2017, 13, e1006771. [Google Scholar] [CrossRef] [PubMed]
- Udugama, M.; Sanij, E.; Voon, H.; Son, J.; Hii, L.; Henson, J.; Chan, F.L.; Chang, F.T.M.; Liu, Y.; Pearson, R.B.; et al. Ribosomal DNA copy loss and repeat instability in ATRX-mutated cancers. Proc. Natl. Acad. Sci. USA 2018, 115, 4737–4742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valori, V.; Tus, K.; Laukaitis, C.; Harris, D.T.; Lebeau, L.; Maggert, K.A. Human rDNA copy number is unstable in metastatic breast cancers. Epigenetics 2020, 15, 85–106. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Lemos, B. Ribosomal DNA copy number amplification and loss in human cancers is linked to tumor genetic con-text, nucleolus activity, and proliferation. PLOS Genet. 2017, 13, e1006994. [Google Scholar] [CrossRef]
- Nelson, J.O.; Watase, G.; Warsinger-Pepe, N.; Yamashita, Y.M. Mechanisms of rDNA Copy Number Maintenance. Trends Genet. 2019, 35, 734–742. [Google Scholar] [CrossRef]
- Warmerdam, D.O.; Wolthuis, R. Keeping ribosomal DNA intact: A repeating challenge. Chromosom. Res. 2019, 27, 57–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindström, M.S.; Jurada, D.; Bursac, S.; Oršolić, I.; Bartek, J.; Volarevic, S. Nucleolus as an emerging hub in maintenance of genome stability and cancer pathogenesis. Oncogene 2018, 37, 2351–2366. [Google Scholar] [CrossRef] [PubMed]
- Santos-Pereira, J.M.; Aguilera, A. R loops: New modulators of genome dynamics and function. Nat. Rev. Genet. 2015, 16, 583–597. [Google Scholar] [CrossRef] [PubMed]
- De Magis, A.; Manzo, S.G.; Russo, M.; Marinello, J.; Morigi, R.; Sordet, O.; Capranico, G. DNA damage and genome instability by G-quadruplex ligands are mediated by R loops in human cancer cells. Proc. Natl. Acad. Sci. USA 2019, 116, 816–825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ginno, P.A.; Lott, P.; Christensen, H.C.; Korf, I.; Chédin, F. R-Loop Formation Is a Distinctive Characteristic of Unmethylated Human CpG Island Promoters. Mol. Cell 2012, 45, 814–825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akamatsu, Y.; Kobayashi, T. The Human RNA Polymerase I Transcription Terminator Complex Acts as a Replication Fork Barrier That Coordinates the Progress of Replication with rRNA Transcription Activity. Mol. Cell. Biol. 2015, 35, 1871–1881. [Google Scholar] [CrossRef] [Green Version]
- Killen, M.W.; Stults, D.M.; Adachi, N.; Hanakahi, L.; Pierce, A.J. Loss of Bloom syndrome protein destabilizes human gene cluster architecture. Hum. Mol. Genet. 2009, 18, 3417–3428. [Google Scholar] [CrossRef] [Green Version]
- Stults, D.M.; Killen, M.W.; Williamson, E.P.; Hourigan, J.S.; Vargas, H.D.; Arnold, S.M.; Moscow, J.A.; Pierce, A.J. Human rRNA Gene Clusters Are Recombinational Hotspots in Cancer. Cancer Res. 2009, 69, 9096–9104. [Google Scholar] [CrossRef] [Green Version]
- Tchurikov, N.A.; Fedoseeva, D.M.; Sosin, D.V.; Snezhkina, A.V.; Melnikova, N.V.; Kudryavtseva, A.V.; Kravatsky, Y.V.; Kretova, O.V. Hot spots of DNA double-strand breaks and genomic contacts of human rDNA units are involved in epigenet-ic regulation. J. Mol. Cell Biol. 2015, 7, 366–382. [Google Scholar] [CrossRef]
- MacLeod, R.A.; Spitzer, D.; Bar-Am, I.; Sylvester, J.E.; Kaufmann, M.; Wernich, A.; Drexler, H.G. Karyotypic dissection of Hodgkin’s disease cell lines reveals ectopic subtelomeres and ribosomal DNA at sites of multiple jumping translocations and genomic amplification. Leukemia 2000, 14, 1803–1814. [Google Scholar] [CrossRef] [Green Version]
- Feng, L.; Du, J.; Yao, C.; Jiang, Z.; Li, T.; Zhang, Q.; Guo, X.; Yu, M.; Xia, H.; Shi, L.; et al. Ribosomal DNA copy number is associated with P53 status and levels of heavy metals in gastrectomy specimens from gastric cancer patients. Environ. Int. 2020, 138, 105593. [Google Scholar] [CrossRef] [PubMed]
- Hosgood, H.D.; Hu, W.; Rothman, N.; Klugman, M.; Weinstein, S.J.; Virtamo, J.R.; Albanes, D.; Cawthon, R.; Lan, Q. Variation in ribosomal DNA copy number is associated with lung cancer risk in a prospective cohort study. Carcinogenesis 2019, 40, 975–978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korsholm, L.M.; Gál, Z.; Nieto, B.; Quevedo, O.; Boukoura, S.; Lund, C.C.; Larsen, D.H. Recent advances in the nucleolar responses to DNA double-strand breaks. Nucleic Acids Res. 2020, 48, 9449–9461. [Google Scholar] [CrossRef]
- Harding, S.; Boiarsky, J.A.; Greenberg, R.A. ATM Dependent Silencing Links Nucleolar Chromatin Reorganization to DNA Damage Recognition. Cell Rep. 2015, 13, 251–259. [Google Scholar] [CrossRef] [Green Version]
- Larsen, D.H.; Hari, F.J.; Clapperton, J.A.; Gwerder, M.; Gutsche, K.; Altmeyer, M.; Jungmichel, S.; Toledo, L.; Fink, D.; Rask, M.-B.; et al. The NBS1–Treacle complex controls ribosomal RNA transcription in response to DNA damage. Nat. Cell Biol. 2014, 16, 792–803. [Google Scholar] [CrossRef] [Green Version]
- Van Sluis, M.; McStay, B. A localized nucleolar DNA damage response facilitates recruitment of the homology-directed repair machinery independent of cell cycle stage. Genes Dev. 2015, 29, 1151–1163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berkovich, E.M.R.J., Jr.; Kastan, M.B. Roles of ATM and NBS1 in chromatin structure modulation and DNA double-strand break repair. Nat. Cell Biol. 2007, 9, 683–690. [Google Scholar] [CrossRef]
- Kruhlak, M.; Crouch, E.E.; Orlov, M.; Montaño, C.; Gorski, S.; Nussenzweig, A.; Misteli, T.; Phair, R.D.; Casellas, R. The ATM repair pathway inhibits RNA polymerase I transcription in response to chromosome breaks. Nat. Cell Biol. 2007, 447, 730–734. [Google Scholar] [CrossRef] [PubMed]
- Ciccia, A.; Huang, J.-W.; Izhar, L.; Sowa, M.E.; Harper, J.W.; Elledge, S.J. Treacher Collins syndrome TCOF1 protein cooperates with NBS1 in the DNA damage response. Proc. Natl. Acad. Sci. USA 2014, 111, 18631–18636. [Google Scholar] [CrossRef] [Green Version]
- Korsholm, L.M.; Gál, Z.; Lin, L.; Quevedo, O.; Ahmad, D.A.; Dulina, E.; Luo, Y.; Bartek, J.; Larsen, D.H. Double-strand breaks in ribosomal RNA genes activate a distinct signaling and chromatin response to facilitate nucleolar restructuring and repair. Nucleic Acids Res. 2019, 47, 8019–8035. [Google Scholar] [CrossRef] [Green Version]
- Mooser, C.; Symeonidou, I.-E.; Leimbacher, P.-A.; Ribeiro, A.; Shorrocks, A.-M.K.; Jungmichel, S.; Larsen, S.C.; Knechtle, K.; Jasrotia, A.; Zurbriggen, D.; et al. Treacle controls the nucleolar response to rDNA breaks via TOPBP1 recruitment and ATR activation. Nat. Commun. 2020, 11, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.-T.; Xu, B.; Kastan, M.B. Involvement of the cohesin protein, Smc1, in Atm-dependent and independent responses to DNA damage. Genes Dev. 2002, 16, 560–570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pefani, D.E.; Tognoli, M.L.; Pirincci Ercan, D.; Gorgoulis, V.; O’Neill, E. MST2 kinase suppresses rDNA transcription in re-sponse to DNA damage by phosphorylating nucleolar histone H2B. EMBO J. 2018, 37, e98760. [Google Scholar] [CrossRef]
- Siebenwirth, C.; Greubel, C.; Drexler, G.A.; Reindl, J.; Walsh, D.W.M.; Schwarz, B.; Sammer, M.; Baur, I.; Pospiech, H.; Schmid, T.E.; et al. Local inhibition of rRNA transcription without nucleolar segregation after targeted ion irradiation of the nucleolus. J. Cell Sci. 2019, 132, jcs232181. [Google Scholar] [CrossRef] [Green Version]
- Warmerdam, D.O.; van den Berg, J.; Medema, R.H. Breaks in the 45S rDNA Lead to Recombination-Mediated Loss of Repeats. Cell Rep 2016, 14, 2519–2527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Sluis, M.; McStay, B. Nucleolar reorganization in response to rDNA damage. Curr. Opin. Cell Biol. 2017, 46, 81–86. [Google Scholar] [CrossRef]
- Jarboui, M.A.; Wynne, K.; Elia, G.; Hall, W.W.; Gautier, V.W. Proteomic profiling of the human T-cell nucleolus. Mol. Immunol. 2011, 49, 441–452. [Google Scholar] [CrossRef]
- Boisvert, F.-M.; Van Koningsbruggen, S.; Navascués, J.; Lamond, A.I. The multifunctional nucleolus. Nat. Rev. Mol. Cell Biol. 2007, 8, 574–585. [Google Scholar] [CrossRef]
- Couté, Y.; Burgess, J.A.; Diaz, J.-J.; Chichester, C.; Lisacek, F.; Greco, A.; Sanchez, J.-C. Deciphering the human nucleolar proteome. Mass Spectrom. Rev. 2006, 25, 215–234. [Google Scholar] [CrossRef]
- Andersen, J.; Lam, Y.W.; Leung, A.; Ong, S.-E.; Lyon, C.E.; Lamond, A.; Mann, M. Nucleolar proteome dynamics. Nat. Cell Biol. 2005, 433, 77–83. [Google Scholar] [CrossRef] [PubMed]
- Moore, H.M.; Bai, B.; Boisvert, F.M.; Latonen, L.; Rantanen, V.; Simpson, J.C.; Pepperkok, R.; Lamond, A.I.; Laiho, M. Quan-titative proteomics and dynamic imaging of the nucleolus reveal distinct responses to UV and ionizing radiation. Mol. Cell Proteom. 2011, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogawa, L.M.; Baserga, S.J. Crosstalk between the nucleolus and the DNA damage response. Mol. BioSyst. 2017, 13, 443–455. [Google Scholar] [CrossRef]
- Rancourt, A.; Satoh, M.S. Delocalization of nucleolar poly (ADP-ribose) polymerase-1 to the nucleoplasm and its novel link to cellular sensitivity to DNA damage. DNA Repair 2009, 8, 286–297. [Google Scholar] [CrossRef]
- Pascal, J.M. The comings and goings of PARP-1 in response to DNA damage. DNA Repair 2018, 71, 177–182. [Google Scholar] [CrossRef]
- Sfeir, A.; Symington, L.S. Microhomology-Mediated End Joining: A Back-up Survival Mechanism or Dedicated Pathway? Trends Biochem. Sci. 2015, 40, 701–714. [Google Scholar] [CrossRef] [Green Version]
- Black, S.J.; Ozdemir, A.Y.; Kashkina, E.; Kent, T.; Rusanov, T.; Ristic, D.; Shin, Y.; Suma, A.; Hoang, T.; Chandramouly, G.; et al. Molecular basis of microhomology-mediated end-joining by purified full-length Polθ. Nat. Commun. 2019, 10, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Guetg, C.; Scheifele, F.; Rosenthal, F.; Hottiger, M.; Santoro, R. Inheritance of Silent rDNA Chromatin Is Mediated by PARP1 via Noncoding RNA. Mol. Cell 2012, 45, 790–800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guetg, C.; Santoro, R. Noncoding RNAs link PARP1 to heterochromatin. Cell Cycle 2012, 11, 2217–2218. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.-S.; Camacho, C.; Nagari, A.; Malladi, V.; Challa, S.; Kraus, W.L. Activation of PARP-1 by snoRNAs Controls Ribosome Biogenesis and Cell Growth via the RNA Helicase DDX21. Mol. Cell 2019, 75, 1270–1285.e14. [Google Scholar] [CrossRef]
- Veith, S.; Schink, A.; Engbrecht, M.; Mack, M.; Rank, L.; Rossatti, P.; Hakobyan, M.; Goly, D.; Hefele, T.; Frensch, M.; et al. PARP1 regulates DNA damage-induced nucleolar-nucleoplasmic shuttling of WRN and XRCC1 in a toxicant and protein-specific manner. Sci. Rep. 2019, 9, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.; Wu, W.; Togashi, Y.; Liang, W.; Miyoshi, Y.; Ohta, T. HERC2 inactivation abrogates nucleolar localization of RecQ helicases BLM and WRN. Sci. Rep. 2021, 11, 1–13. [Google Scholar] [CrossRef]
- Fung, H.; Demple, B. A Vital Role for Ape1/Ref1 Protein in Repairing Spontaneous DNA Damage in Human Cells. Mol. Cell 2005, 17, 463–470. [Google Scholar] [CrossRef] [PubMed]
- Lirussi, L.; Antoniali, G.; Vascotto, C.; D’Ambrosio, C.; Poletto, M.; Romanello, M.; Marasco, D.; Leone, M.; Quadrifoglio, F.; Bhakat, K.K.; et al. Nucleolar accumulation of APE1 depends on charged lysine residues that undergo acetylation upon geno-toxic stress and modulate its BER activity in cells. Mol. Biol. Cell 2012, 23, 4079–4096. [Google Scholar] [CrossRef]
- Vascotto, C.; Fantini, D.; Romanello, M.; Cesaratto, L.; Deganuto, M.; Leonardi, A.; Radicella, J.P.; Kelley, M.R.; D’Ambrosio, C.; Scaloni, A.; et al. APE1/Ref-1 Interacts with NPM1 within Nucleoli and Plays a Role in the rRNA Quality Control Process. Mol. Cell. Biol. 2009, 29, 1834–1854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsuoka, S.; Ballif, B.A.; Smogorzewska, A.; McDonald, E.R.; Hurov, K.E.; Luo, J.; Bakalarski, C.; Zhao, Z.; Solimini, N.; Lerenthal, Y.; et al. ATM and ATR Substrate Analysis Reveals Extensive Protein Networks Responsive to DNA Damage. Science 2007, 316, 1160–1166. [Google Scholar] [CrossRef] [Green Version]
- Rubbi, C.P.; Milner, J. Disruption of the nucleolus mediates stabilization of p53 in response to DNA damage and other stress-es. EMBO J. 2003, 22, 6068–6077. [Google Scholar] [CrossRef] [Green Version]
- Pestov, D.G.; Strezoska, Ž.; Lau, L.F. Evidence of p53-Dependent Cross-Talk between Ribosome Biogenesis and the Cell Cycle: Effects of Nucleolar Protein Bop1 on G1/S Transition. Mol. Cell. Biol. 2001, 21, 4246–4255. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Deisenroth, C.; Zhang, Y. RP-MDM2-p53 Pathway: Linking Ribosomal Biogenesis and Tumor Surveillance. Trends Cancer 2016, 2, 191–204. [Google Scholar] [CrossRef] [Green Version]
- Gjerset, R.A.; Bandyopadhyay, K. Regulation of p14ARF Through Subnuclear Compartmentalization. Cell Cycle 2006, 5, 686–690. [Google Scholar] [CrossRef] [Green Version]
- Freedman, D.A.; Wu, L.; Levine, A.J. Functions of the MDM2 oncoprotein. Cell. Mol. Life Sci. 1999, 55, 96–107. [Google Scholar] [CrossRef]
- Bernardi, R.; Scaglioni, P.P.; Bergmann, S.; Horn, H.F.; Vousden, K.H.; Pandolfi, P.P. PML regulates p53 stability by seques-tering Mdm2 to the nucleolus. Nat. Cell Biol. 2004, 6, 665–672. [Google Scholar] [CrossRef] [PubMed]
- Pelletier, J.; Thomas, G.; Volarević, S. Ribosome biogenesis in cancer: New players and therapeutic avenues. Nat. Rev. Cancer 2018, 18, 51–63. [Google Scholar] [CrossRef]
- Bruno, P.M.; Liu, Y.; Park, G.Y.; Murai, J.; Koch, C.E.; Eisen, T.J.; Pritchard, J.R.; Pommier, Y.; Lippard, S.J.; Hemann, M.T. A subset of platinum-containing chemotherapeutic agents kills cells by inducing ribosome biogenesis stress. Nat. Med. 2017, 23, 461–471. [Google Scholar] [CrossRef]
- Quin, J.E.; Devlin, J.R.; Cameron, D.; Hannan, K.M.; Pearson, R.B.; Hannan, R.D. Targeting the nucleolus for cancer intervention. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2014, 1842, 802–816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khot, A.; Brajanovski, N.; Cameron, D.P.; Hein, N.; Maclachlan, K.H.; Sanij, E.; Lim, J.; Soong, J.; Link, E.; Blombery, P.; et al. First-in-Human RNA Polymerase I Transcription Inhibitor CX-5461 in Patients with Advanced Hematologic Cancers: Results of a Phase I Dose-Escalation Study. Cancer Discov. 2019, 9, 1036–1049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peltonen, K.D.; Colis, L.; Liu, H.; Trivedi, R.; Moubarek, M.S.; Moore, H.M.; Bai, B.; Rudek, M.A.; Bieberich, C.; Laiho, M. A Targeting Modality for Destruction of RNA Polymerase I that Possesses Anticancer Activity. Cancer Cell 2014, 25, 77–90. [Google Scholar] [CrossRef] [Green Version]
- Quin, J.; Chan, K.T.; Devlin, J.R.; Cameron, D.P.; Diesch, J.; Cullinane, C.; Ahern, J.; Khot, A.; Hein, N.; George, A.J.; et al. Inhibition of RNA polymerase I transcription initiation by CX-5461 activates non-canonical ATM/ATR signaling. Oncotarget 2016, 7, 49800–49818. [Google Scholar] [CrossRef] [Green Version]
- Wei, T.; Najmi, S.M.; Liu, H.; Peltonen, K.; Kucerova, A.; Schneider, D.A.; Laiho, M. Small-Molecule Targeting of RNA Pol-ymerase I Activates a Conserved Transcription Elongation Checkpoint. Cell Rep. 2018, 23, 404–414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferreira, R.; Schneekloth, J.J.S.; Panov, K.I.; Hannan, K.M.; Hannan, R.D. Targeting the RNA Polymerase I Transcription for Cancer Therapy Comes of Age. Cells 2020, 9, 266. [Google Scholar] [CrossRef] [Green Version]
- Drygin, D.; Lin, A.; Bliesath, J.; Ho, C.B.; O’Brien, S.E.; Proffitt, C.; Omori, M.; Haddach, M.; Schwaebe, M.K.; Siddiqui-Jain, A.; et al. Targeting RNA Polymerase I with an Oral Small Molecule CX-5461 Inhibits Ribosomal RNA Synthesis and Solid Tumor Growth. Cancer Res. 2011, 71, 1418–1430. [Google Scholar] [CrossRef] [Green Version]
- Haddach, M.; Schwaebe, M.K.; Michaux, J.; Nagasawa, J.; O’Brien, S.E.; Whitten, J.P.; Pierre, F.; Kerdoncuff, P.; Darjania, L.; Stansfield, R.; et al. Discovery of CX-5461, the First Direct and Selective Inhibitor of RNA Polymerase I, for Cancer Therapeutics. ACS Med. Chem. Lett. 2012, 3, 602–606. [Google Scholar] [CrossRef] [Green Version]
- Bywater, M.J.; Poortinga, G.; Sanij, E.; Hein, N.; Peck, A.; Cullinane, C.; Wall, M.; Cluse, L.; Drygin, D.; Anderes, K.; et al. Inhibition of RNA Polymerase I as a Therapeutic Strategy to Promote Cancer-Specific Activation of p53. Cancer Cell 2012, 22, 51–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rebello, R.J.; Kusnadi, E.; Cameron, D.P.; Pearson, H.B.; Lesmana, A.; Devlin, J.R.; Drygin, D.; Clark, A.K.; Porter, L.; Pedersen, J.; et al. The Dual Inhibition of RNA Pol I Transcription and PIM Kinase as a New Therapeutic Approach to Treat Advanced Prostate Cancer. Clin. Cancer Res. 2016, 22, 5539–5552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hein, N.; Cameron, D.P.; Hannan, K.M.; Nguyen, N.-Y.N.; Fong, C.Y.; Sornkom, J.; Wall, M.; Pavy, M.; Cullinane, C.; Diesch, J.; et al. Inhibition of Pol I transcription treats murine and human AML by targeting the leukemia-initiating cell population. Blood 2017, 129, 2882–2895. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Di Antonio, M.; McKinney, S.; Mathew, V.; Ho, B.; O’Neil, N.J.; Santos, N.D.; Silvester, J.; Wei, V.; Garcia, J.; et al. CX-5461 is a DNA G-quadruplex stabilizer with selective lethality in BRCA1/2 deficient tumours. Nat. Commun. 2017, 8. [Google Scholar] [CrossRef]
- Sanij, E.; Hannan, K.M.; Xuan, J.; Yan, S.; Ahern, J.E.; Trigos, A.S.; Brajanovski, N.; Son, J.; Chan, K.T.; Kondrashova, O.; et al. CX-5461 activates the DNA damage response and demonstrates therapeutic efficacy in high-grade serous ovarian cancer. Nat. Commun. 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Cornelison, R.; Dobbin, Z.C.; Katre, A.A.; Jeong, D.H.; Zhang, Y.; Chen, D.; Petrova, Y.; Llaneza, D.C.; Steg, A.D.; Parsons, L.; et al. Targeting RNA-Polymerase I in Both Chemosensitive and Chemoresistant Populations in Epithelial Ovarian Cancer. Clin. Cancer Res. 2017, 23, 6529–6540. [Google Scholar] [CrossRef] [Green Version]
- Negi, S.S.; Brown, P. rRNA synthesis inhibitor, CX-5461, activates ATM/ATR pathway in acute lymphoblastic leukemia, arrests cells in G2 phase and induces apoptosis. Oncotarget 2015, 6, 18094–18104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hilton, J.; Gelmon, K.; Cescon, D.; Tinker, A.; Jonker, D.; Goodwin, R.; Laurie, S.; Hansen, A.; Aparicio, S.; Soong, J.; et al. Abstract: Canadian cancer trials group trial IND.231: A phase 1 trial evaluating CX-5461, a novel first-in-class G-quadruplex stabilizer in patients with advanced solid tumors enriched for DNA-repair deficiencies. In: Proceedings of the 2019 San Antonio Breast Cancer Symposium; 2019 Dec 10-14; San Antonio, TX. Philadelphia (PA): AACR. Cancer Res 2020, 80 (Suppl. 4). [Google Scholar] [CrossRef]
- Li, X.; Li, L.; Li, Y.; Zhao, J.; Fan, S.; Wang, L. CX-5461 induces autophagy and inhibits tumor growth via mammalian target of rapamycin-related signaling pathways in osteosarcoma. OncoTargets Ther. 2016, 9, 5985–5997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drygin, D.; Rice, W.G.; Grummt, I. The RNA Polymerase I Transcription Machinery: An Emerging Target for the Treatment of Cancer. Annu. Rev. Pharmacol. Toxicol. 2010, 50, 131–156. [Google Scholar] [CrossRef] [PubMed]
- Goodfellow, S.J.; Zomerdijk, J.C.B.M. Basic Mechanisms in RNA Polymerase I Transcription of the Ribosomal RNA Genes. Prokaryotic Cytoskelet. 2013, 61, 211–236. [Google Scholar] [CrossRef] [Green Version]
- Tuan, J.C.; Zhai, W.; Comai, L. Recruitment of TATA-Binding Protein–TAFI Complex SL1 to the Human Ribosomal DNA Promoter Is Mediated by the Carboxy-Terminal Activation Domain of Upstream Binding Factor (UBF) and Is Regulated by UBF Phosphorylation. Mol. Cellular Biol. 1999, 19, 2872–2879. [Google Scholar] [CrossRef] [Green Version]
- Miller, G.; Panov, K.I.; Friedrich, J.K.; Trinkle-Mulcahy, L.; Lamond, A.I.; Zomerdijk, J.C. hRRN3 is essential in the SL1-mediated recruitment of RNA Polymerase I to rRNA gene promoters. EMBO J. 2001, 20, 1373–1382. [Google Scholar] [CrossRef] [Green Version]
- Bell, S.P.; Learned, R.M.; Jantzen, H.M.; Tjian, R. Functional cooperativity between transcription factors UBF1 and SL1 mediates human ribosomal RNA synthesis. Science 1988, 241, 1192–1197. [Google Scholar] [CrossRef] [PubMed]
- Friedrich, J.K.; Panov, K.; Čabart, P.; Russell, J.; Zomerdijk, J.C.B.M. TBP-TAF Complex SL1 Directs RNA Polymerase I Pre-initiation Complex Formation and Stabilizes Upstream Binding Factor at the rDNA Promoter. J. Biol. Chem. 2005, 280, 29551–29558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ray, S.; Panova, T.; Miller, G.; Volkov, A.; Porter, A.C.G.; Russell, J.; Panov, K.I.; Zomerdijk, J.C.B.M. Topoisomerase IIα promotes activation of RNA polymerase I transcription by facilitating pre-initiation complex formation. Nat. Commun. 2013, 4, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panov, K.I.; Friedrich, J.K.; Russell, J.; Zomerdijk, J.C.B.M. UBF activates RNA polymerase I transcription by stimulating promoter escape. EMBO J. 2006, 25, 3310–3322. [Google Scholar] [CrossRef] [Green Version]
- Ide, S.; Imai, R.; Ochi, H.; Maeshima, K. Transcriptional suppression of ribosomal DNA with phase separation. Sci. Adv. 2020, 6, eabb5953. [Google Scholar] [CrossRef]
- Mars, J.-C.; Tremblay, M.G.; Valere, M.; Sibai, D.S.; Sabourin-Felix, M.; Lessard, F.; Moss, T. The chemotherapeutic agent CX-5461 irreversibly blocks RNA polymerase I initiation and promoter release to cause nucleolar disruption, DNA damage and cell inviability. NAR Cancer 2020, 2, zcaa032. [Google Scholar] [CrossRef]
- Bruno, P.M.; Lu, M.; Dennis, K.A.; Inam, H.; Moore, C.J.; Sheehe, J.; Elledge, S.J.; Hemann, M.T.; Pritchard, J.R. The primary mechanism of cytotoxicity of the chemotherapeutic agent CX-5461 is topoisomerase II poisoning. Proc. Natl. Acad. Sci. USA 2020, 117, 4053–4060. [Google Scholar] [CrossRef]
- Brill, S.J.; Dinardo, S.; Voelkel-meiman, K.; Sternglanz, R. Need for DNA topoisomerase activity as a swivel for DNA replica-tion for transcription of ribosomal RNA. Nature 1987, 326, 414–416. [Google Scholar] [CrossRef]
- Wang, J.C. Cellular roles of DNA topoisomerases: A molecular perspective. Nat. Rev. Mol. Cell Biol. 2002, 3, 430–440. [Google Scholar] [CrossRef] [PubMed]
- Nitiss, J.L. DNA topoisomerase II and its growing repertoire of biological functions. Nat. Rev. Cancer 2009, 9, 327–337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pommier, Y.; Sun, Y.; Huang, S.-Y.N.; Nitiss, J. Roles of eukaryotic topoisomerases in transcription, replication and genomic stability. Nat. Rev. Mol. Cell Biol. 2016, 17, 703–721. [Google Scholar] [CrossRef] [PubMed]
- Champoux, J.J. DNA Topoisomerases: Structure, Function, and Mechanism. Annu. Rev. Biochem. 2001, 70, 369–413. [Google Scholar] [CrossRef] [Green Version]
- Wilstermann, A.; Osheroff, N. Stabilization of Eukaryotic Topoisomerase II-DNA Cleavage Complexes. Curr. Topics Med. Chem. 2005, 3, 321–338. [Google Scholar] [CrossRef]
- Pommier, Y.; Leo, E.; Zhang, H.; Marchand, C. DNA Topoisomerases and Their Poisoning by Anticancer and Antibacterial Drugs. Chem. Biol. 2010, 17, 421–433. [Google Scholar] [CrossRef] [Green Version]
- Kawatani, M.; Takayama, H.; Muroi, M.; Kimura, S.; Maekawa, T.; Osada, H. Identification of a Small-Molecule Inhibitor of DNA Topoisomerase II by Proteomic Profiling. Chem. Biol. 2011, 18, 743–751. [Google Scholar] [CrossRef] [Green Version]
- Pipier, A.; Bossaert, M.; Riou, J.F.; Noirot, C.; Nguyễn, L.T.; Serre, R.F.; Bouchez, O.; Defrancq, E.; Calsou, P.; Britton, S.; et al. Transcription-associated topoisomerase activities control DNA-breaks production by G-quadruplex ligands. bioRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Yan, S.; Xuan, J.; Brajanovski, N.; Tancock, M.R.C.; Madhamshettiwar, P.B.; Simpson, K.J.; Ellis, S.; Kang, J.; Cullinane, C.; Sheppard, K.E.; et al. The RNA polymerase I transcription inhibitor CX-5461 cooperates with topoisomerase 1 inhibition by enhancing the DNA damage response in homologous recombination-proficient high-grade serous ovarian cancer. Br. J. Cancer 2021, 124, 616–627. [Google Scholar] [CrossRef] [PubMed]
- Bossaert, M.; Pipier, A.; Riou, J.F.; Noirot, C.; Nguyen, L.T.; Serre, R.F.; Bouchez, O.; Defrancq, E.; Calsou, P.; Britton, S.; et al. Transcription-associated topoisomerase 2alpha (TOP2A) activity is a major effector of cytotoxicity induced by G-quadruplex ligands. Elife 2021, 10. [Google Scholar] [CrossRef] [PubMed]
- Son, J.; Hannan, K.M.; Poortinga, G.; Hein, N.; Cameron, D.P.; Ganley, A.R.D.; Sheppard, K.E.; Pearson, R.B.; Hannan, R.D.; Sanij, E. rDNA Chromatin Activity Status as a Biomarker of Sensitivity to the RNA Polymerase I Transcription Inhibitor CX-5461. Front. Cell Dev. Biol. 2020, 8, 568. [Google Scholar] [CrossRef]
- Ruggero, D.; Pandolfi, P.P. Does the ribosome translate cancer? Nat. Rev. Cancer 2003, 3, 179–192. [Google Scholar] [CrossRef]
- Negi, S.S.; Brown, P. Transient rRNA synthesis inhibition with CX-5461 is sufficient to elicit growth arrest and cell death in acute lymphoblastic leukemia cells. Oncotarget 2015, 6, 34846–34858. [Google Scholar] [CrossRef] [Green Version]
- Lawrence, M.; Porter, L.; Choo, N.; Pook, J.; Grummet, J.; Pezaro, C.J.; Sandhu, S.; Ramm, S.; Luu, J.; Bakshi, A.; et al. CX-5461 sensitises DNA damage repair proficient castrate-resistant prostate cancer to PARP inhibition. Mol. Cancer Ther. 2021. Accepted on 17 April 2021. [Google Scholar]
- Muller, M.T.; Pfund, W.P.; Mehta, V.B.; Trask, D.K. Eukaryotic type I topoisomerase is enriched in the nucleolus and catalyti-cally active on ribosomal DNA. EMBO J. 1985, 4, 1237–1243. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xuan, J.; Gitareja, K.; Brajanovski, N.; Sanij, E. Harnessing the Nucleolar DNA Damage Response in Cancer Therapy. Genes 2021, 12, 1156. https://doi.org/10.3390/genes12081156
Xuan J, Gitareja K, Brajanovski N, Sanij E. Harnessing the Nucleolar DNA Damage Response in Cancer Therapy. Genes. 2021; 12(8):1156. https://doi.org/10.3390/genes12081156
Chicago/Turabian StyleXuan, Jiachen, Kezia Gitareja, Natalie Brajanovski, and Elaine Sanij. 2021. "Harnessing the Nucleolar DNA Damage Response in Cancer Therapy" Genes 12, no. 8: 1156. https://doi.org/10.3390/genes12081156