
Autosomal Recessive Retinitis Pigmentosa Associated with Three Novel REEP6 Variants in Chinese Population



Abstract
:1. Introduction
2. Materials and Methods
2.1. Subjects and Clinical Examinations
2.2. Targeted NGS and Sanger Sequencing
2.3. In Silico Analysis
2.4. REEP6 Expression Constructs
2.5. Cell Culture and Transient Transfection
2.6. Real-Time PCR
2.7. Western Blotting
2.8. Immunofluorescence
3. Results
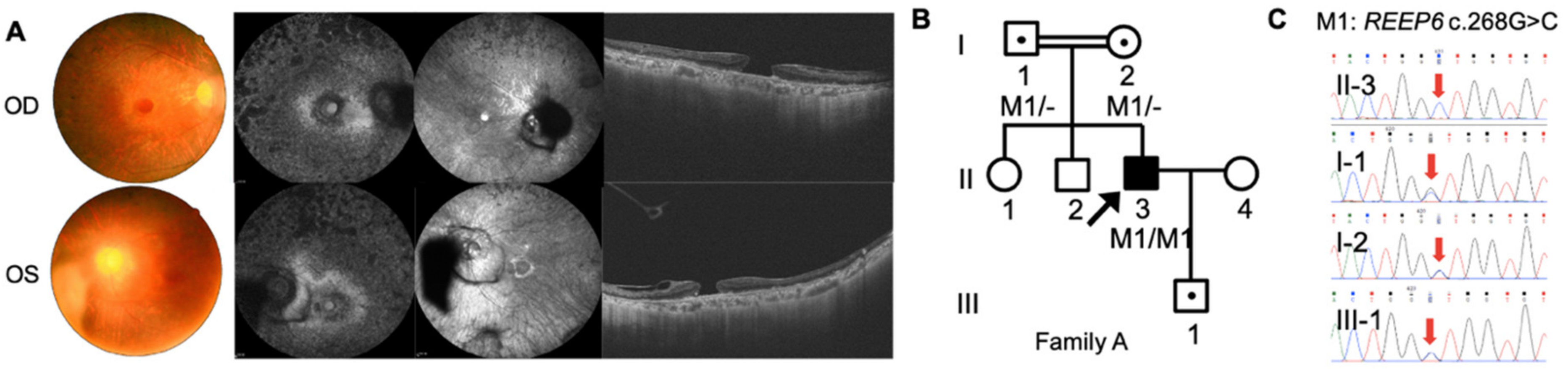
3.1. Clinical Examinations
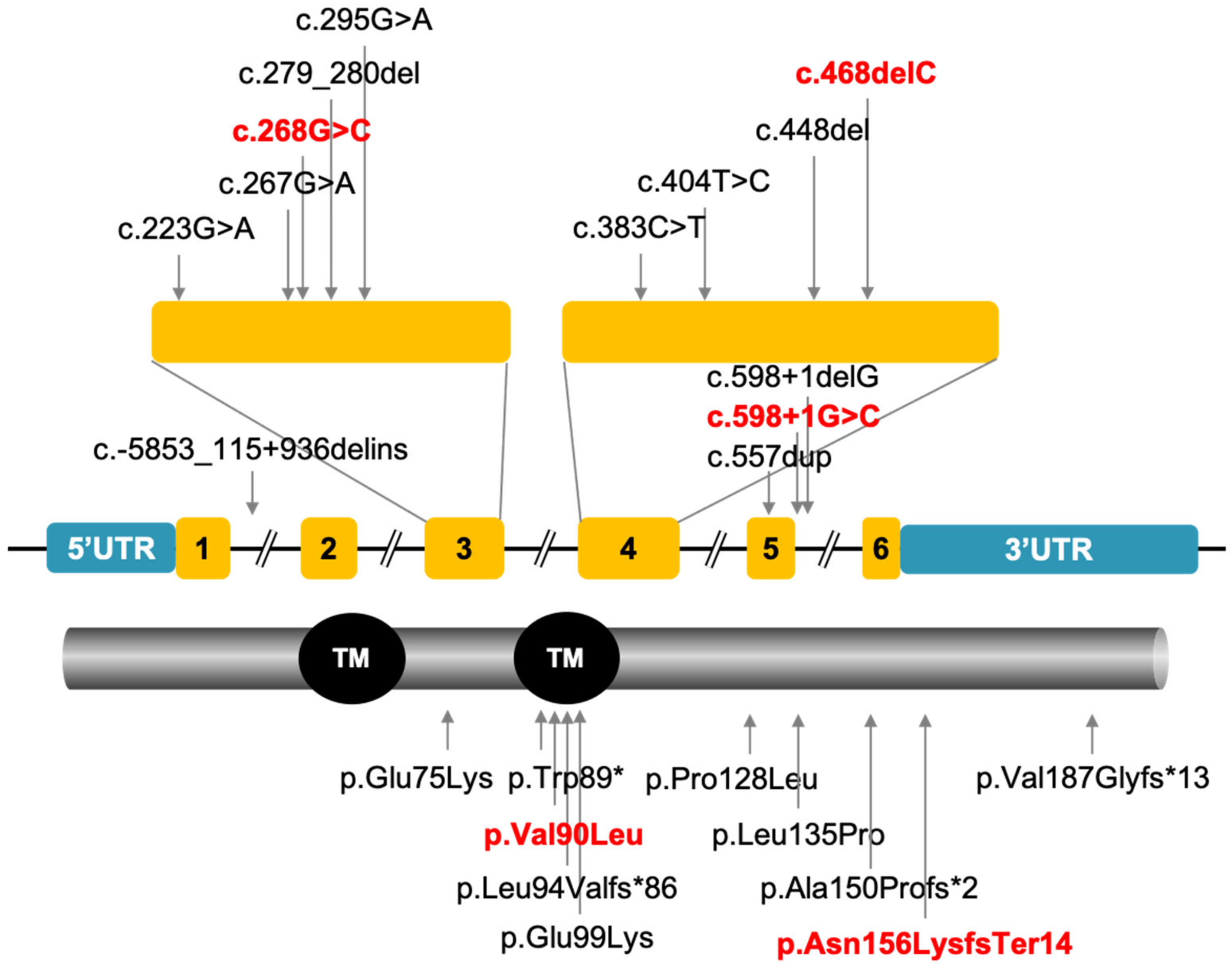
3.2. Mutation Analysis
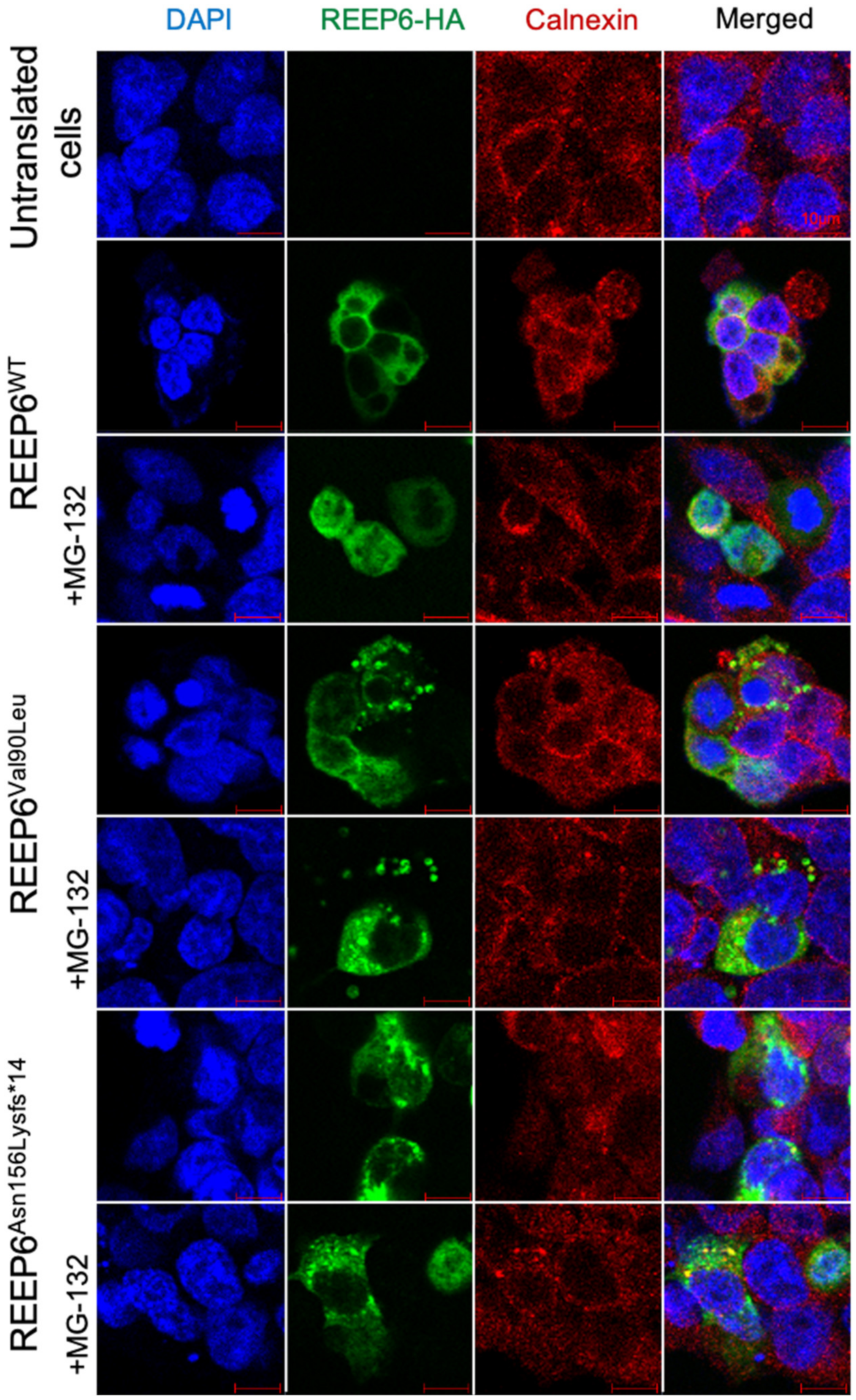
3.3. Variants in REEP6 Affected the Stability of the Encoded Proteins
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Campochiaro, P.A.; Strauss, R.W.; Lu, L.; Hafiz, G.; Wolfson, Y.; Shah, S.M.; Sophie, R.; Mir, T.A.; Scholl, H.P. Is There Excess Oxidative Stress and Damage in Eyes of Patients with Retinitis Pigmentosa. Antioxid. Redox Signal. 2015, 23, 643–648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campochiaro, P.A.; Mir, T.A. The mechanism of cone cell death in Retinitis Pigmentosa. Prog. Retin. Eye Res. 2018, 62, 24–37. [Google Scholar] [CrossRef]
- Björk, S.; Hurt, C.M.; Ho, V.K.; Angelotti, T. REEPs are membrane shaping adapter proteins that modulate specific g protein-coupled receptor trafficking by affecting ER cargo capacity. PLoS ONE 2013, 8, e76366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agrawal, S.A.; Burgoyne, T.; Eblimit, A.; Bellingham, J.; Parfitt, D.A.; Lane, A.; Nichols, R.; Asomugha, C.; Hayes, M.J.; Munro, P.M.; et al. REEP6 deficiency leads to retinal degeneration through disruption of ER homeostasis and protein trafficking. Hum. Mol. Genet. 2017, 26, 2667–2677. [Google Scholar] [CrossRef]
- Veleri, S.; Nellissery, J.; Mishra, B.; Manjunath, S.H.; Brooks, M.J.; Dong, L.; Nagashima, K.; Qian, H.; Gao, C.; Sergeev, Y.V.; et al. REEP6 mediates trafficking of a subset of Clathrin-coated vesicles and is critical for rod photoreceptor function and survival. Hum. Mol. Genet. 2017, 26, 2218–2230. [Google Scholar] [CrossRef] [Green Version]
- Hao, H.; Veleri, S.; Sun, B.; Kim, D.S.; Keeley, P.W.; Kim, J.W.; Yang, H.J.; Yadav, S.P.; Manjunath, S.H.; Sood, R.; et al. Regulation of a novel isoform of Receptor Expression Enhancing Protein REEP6 in rod photoreceptors by bZIP transcription factor NRL. Hum. Mol. Genet. 2014, 23, 4260–4271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arno, G.; Agrawal, S.A.; Eblimit, A.; Bellingham, J.; Xu, M.; Wang, F.; Chakarova, C.; Parfitt, D.A.; Lane, A.; Burgoyne, T.; et al. Mutations in REEP6 Cause Autosomal-Recessive Retinitis Pigmentosa. Am. J. Hum. Genet. 2016, 99, 1305–1315. [Google Scholar] [CrossRef]
- Lin, Y.; Xu, C.L.; Velez, G.; Yang, J.; Tanaka, A.J.; Breazzano, M.P.; Mahajan, V.B.; Sparrow, J.R.; Tsang, S.H. Novel REEP6 gene mutation associated with autosomal recessive retinitis pigmentosa. Doc. Ophthalmol. 2020, 140, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Méjécase, C.; Mohand-Saïd, S.; El Shamieh, S.; Antonio, A.; Condroyer, C.; Blanchard, S.; Letexier, M.; Saraiva, J.P.; Sahel, J.A.; Audo, I.; et al. A novel nonsense variant in REEP6 is involved in a sporadic rod-cone dystrophy case. Clin. Genet. 2018, 93, 707–711. [Google Scholar] [CrossRef] [Green Version]
- Fuster-García, C.; García-García, G.; Jaijo, T.; Blanco-Kelly, F.; Tian, L.; Hakonarson, H.; Ayuso, C.; Aller, E.; Millán, J.M. Expanding the Genetic Landscape of Usher-Like Phenotypes. Investig. Ophthalmol. Vis. Sci. 2019, 60, 4701–4710. [Google Scholar] [CrossRef]
- Zhu, Q.; Rui, X.; Li, Y.; You, Y.; Sheng, X.L.; Lei, B. Identification of Four Novel Variants and Determination of Genotype-Phenotype Correlations for ABCA4 Variants Associated with Inherited Retinal Degenerations. Front. Cell Dev. Biol. 2021, 9, 634843. [Google Scholar] [CrossRef]
- Li, J.; Shi, L.; Zhang, K.; Zhang, Y.; Hu, S.; Zhao, T.; Teng, H.; Li, X.; Jiang, Y.; Ji, L.; et al. VarCards: An integrated genetic and clinical database for coding variants in the human genome. Nucleic Acids Res. 2018, 46, D1039–D1048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hebsgaard, S.M.; Korning, P.G.; Tolstrup, N.; Engelbrecht, J.; Rouzé, P.; Brunak, S. Splice site prediction in Arabidopsis thaliana pre-mRNA by combining local and global sequence information. Nucleic Acids Res. 1996, 24, 3439–3452. [Google Scholar] [CrossRef] [Green Version]
- Reese, M.G.; Eeckman, F.H.; Kulp, D.; Haussler, D. Improved splice site detection in Genie. J. Comput. Biol. 1997, 4, 311–323. [Google Scholar] [CrossRef] [PubMed]
- Sievers, F.; Higgins, D.G. Clustal Omega for making accurate alignments of many protein sequences. Protein Sci. 2018, 27, 135–145. [Google Scholar] [CrossRef] [Green Version]
- Marchler-Bauer, A.; Derbyshire, M.K.; Gonzales, N.R.; Lu, S.; Chitsaz, F.; Geer, L.Y.; Geer, R.C.; He, J.; Gwadz, M.; Hurwitz, D.I.; et al. CDD: NCBI’s conserved domain database. Nucleic Acids Res. 2015, 43, D222–D226. [Google Scholar] [CrossRef] [Green Version]
- Venselaar, H.; Te Beek, T.A.; Kuipers, R.K.; Hekkelman, M.L.; Vriend, G. Protein structure analysis of mutations causing inheritable diseases. An e-Science approach with life scientist friendly interfaces. BMC Bioinform. 2010, 11, 548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garg, V.K.; Avashthi, H.; Tiwari, A.; Jain, P.A.; Ramkete, P.W.; Kayastha, A.M.; Singh, V.K. MFPPI—Multi FASTA ProtParam Interface. Bioinformation 2016, 12, 74–77. [Google Scholar] [CrossRef]
- Bon, L.; Lucchetti, C.; Portolan, F.; Pagan, M. MUPRO: A multipurpose robot. Int. J. Neurosci. 2002, 112, 855–868. [Google Scholar] [CrossRef]
- Capriotti, E.; Fariselli, P.; Casadio, R. I-Mutant2.0: Predicting stability changes upon mutation from the protein sequence or structure. Nucleic Acids Res. 2005, 33, W306–W310. [Google Scholar] [CrossRef] [Green Version]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Qiu, Y.; Shil, P.K.; Zhu, P.; Yang, H.; Verma, A.; Lei, B.; Li, Q. Angiotensin-converting enzyme 2 (ACE2) activator diminazene aceturate ameliorates endotoxin-induced uveitis in mice. Investig. Ophthalmol. Vis. Sci. 2014, 55, 3809–3818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoefele, J.; Sudbrak, R.; Reinhardt, R.; Lehrack, S.; Hennig, S.; Imm, A.; Muerb, U.; Utsch, B.; Attanasio, M.; O’Toole, J.F.; et al. Mutational analysis of the NPHP4 gene in 250 patients with nephronophthisis. Hum. Mutat. 2005, 25, 411. [Google Scholar] [CrossRef] [Green Version]
- Tsang, S.H.; Aycinena, A.; Sharma, T. Ciliopathy: Senior-Løken Syndrome. Adv. Exp. Med. Biol. 2018, 1085, 175–178. [Google Scholar]
- Wang, F.; Wang, H.; Tuan, H.F.; Nguyen, D.H.; Sun, V.; Keser, V.; Bowne, S.J.; Sullivan, L.S.; Luo, H.; Zhao, L.; et al. Next generation sequencing-based molecular diagnosis of retinitis pigmentosa: Identification of a novel genotype-phenotype correlation and clinical refinements. Hum. Genet. 2014, 133, 331–345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fokkema, I.F.; Taschner, P.E.; Schaafsma, G.C.; Celli, J.; Laros, J.F.; den Dunnen, J.T. LOVD v.2.0: The next generation in gene variant databases. Hum. Mutat. 2011, 32, 557–563. [Google Scholar] [CrossRef]
- García-Mata, R.; Bebök, Z.; Sorscher, E.J.; Sztul, E.S. Characterization and dynamics of aggresome formation by a cytosolic GFP-chimera. J. Cell Biol. 1999, 146, 1239–1254. [Google Scholar] [CrossRef] [Green Version]
- Johnston, J.A.; Ward, C.L.; Kopito, R.R. Aggresomes: A cellular response to misfolded proteins. J. Cell Biol. 1998, 143, 1883–1898. [Google Scholar] [CrossRef] [Green Version]
- Coban-Akdemir, Z.; White, J.J.; Song, X.; Jhangiani, S.N.; Fatih, J.M.; Gambin, T.; Bayram, Y.; Chinn, I.K.; Karaca, E.; Punetha, J.; et al. Identifying Genes Whose Mutant Transcripts Cause Dominant Disease Traits by Potential Gain-of-Function Alleles. Am. J. Hum. Genet. 2018, 103, 171–187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindeboom, R.; Vermeulen, M.; Lehner, B.; Supek, F. The impact of nonsense-mediated mRNA decay on genetic disease, gene editing and cancer immunotherapy. Nat. Genet. 2019, 51, 1645–1651. [Google Scholar] [CrossRef]
- Mort, M.; Ivanov, D.; Cooper, D.N.; Chuzhanova, N.A. A meta-analysis of nonsense mutations causing human genetic disease. Hum. Mutat. 2008, 29, 1037–1047. [Google Scholar] [CrossRef] [PubMed]
- Supek, F.; Lehner, B.; Lindeboom, R. To NMD or Not To NMD: Nonsense-Mediated mRNA Decay in Cancer and Other Genetic Diseases. Trends Genet. 2020. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Proband | Diagnosis | Onset Age (y) | Age at Diagnosis (y) | BCVA (OD/OS) | ff-ERG | Funds | Visual Field |
|---|---|---|---|---|---|---|---|
| A: II-3 | RP | 35 | 42 | NLP/LP | non-recordable | bone-spicule deposits bilateral macular holes | NA |
| B: II-4 | RP | 21 | 51 | 0.6/0.4 | severely decreased | bone-spicule deposits thin interdigitation zone | tunnel |
| C: II-4 | RP | 39 | 44 | 0.5/0.4 | NA | cystoid macular edema (OD) narrowed retinal vessels | tunnel |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, L.; Li, Y.; Qin, L.; Wu, Y.; Lei, B. Autosomal Recessive Retinitis Pigmentosa Associated with Three Novel REEP6 Variants in Chinese Population. Genes 2021, 12, 537. https://doi.org/10.3390/genes12040537
Zhang L, Li Y, Qin L, Wu Y, Lei B. Autosomal Recessive Retinitis Pigmentosa Associated with Three Novel REEP6 Variants in Chinese Population. Genes. 2021; 12(4):537. https://doi.org/10.3390/genes12040537
Chicago/Turabian StyleZhang, Lujia, Ya Li, Litao Qin, Yu Wu, and Bo Lei. 2021. "Autosomal Recessive Retinitis Pigmentosa Associated with Three Novel REEP6 Variants in Chinese Population" Genes 12, no. 4: 537. https://doi.org/10.3390/genes12040537