
Hereditary Haemorrhagic Telangiectasia, an Inherited Vascular Disorder in Need of Improved Evidence-Based Pharmaceutical Interventions

Abstract
:1. Introduction
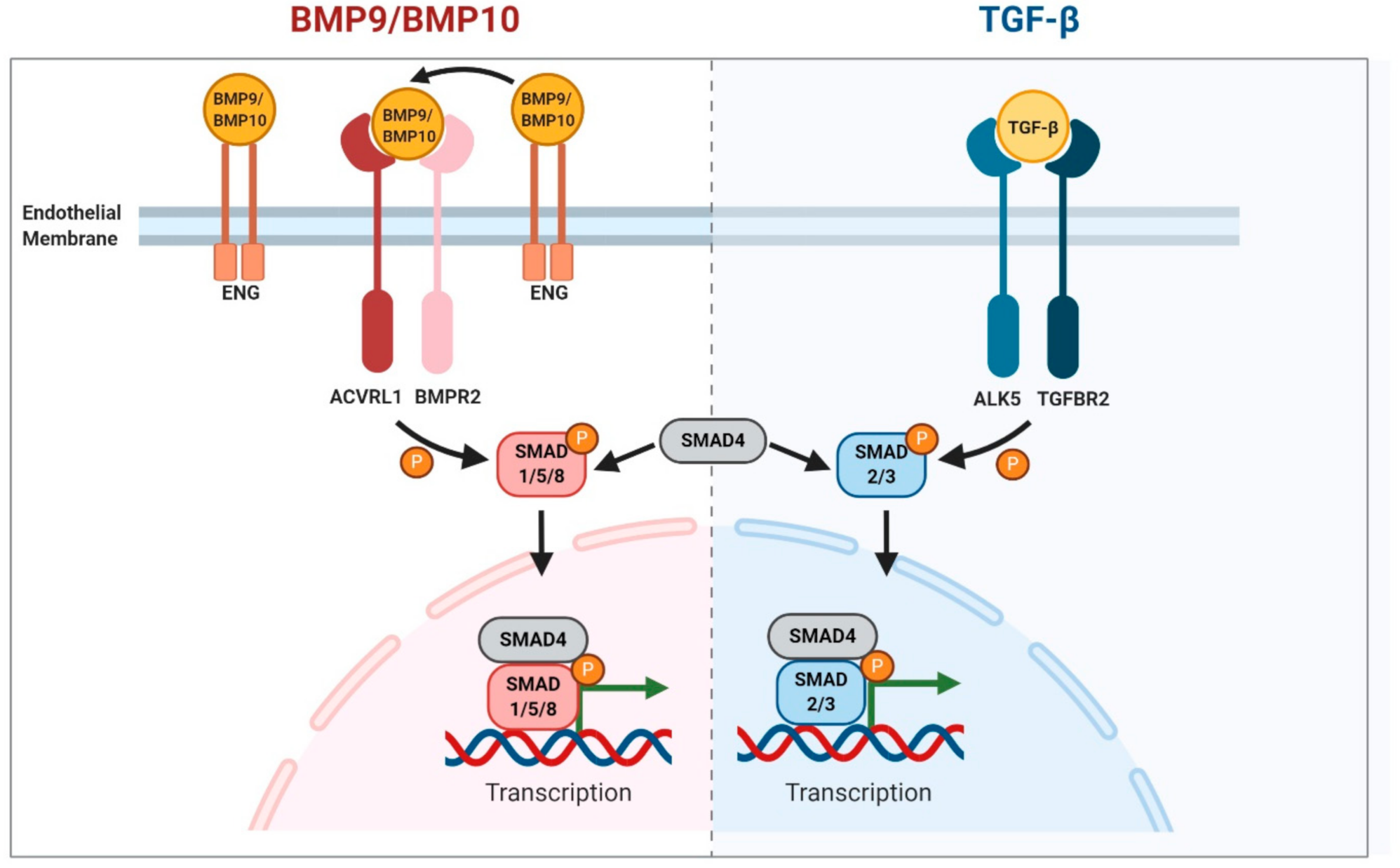
2. BMP9/10 Signalling
3. Aetiology of HHT Disease
4. Overlapping Endothelial Cell Abnormalities in HHT and Spontaneous BAVMs
5. Treatment Options for HHT
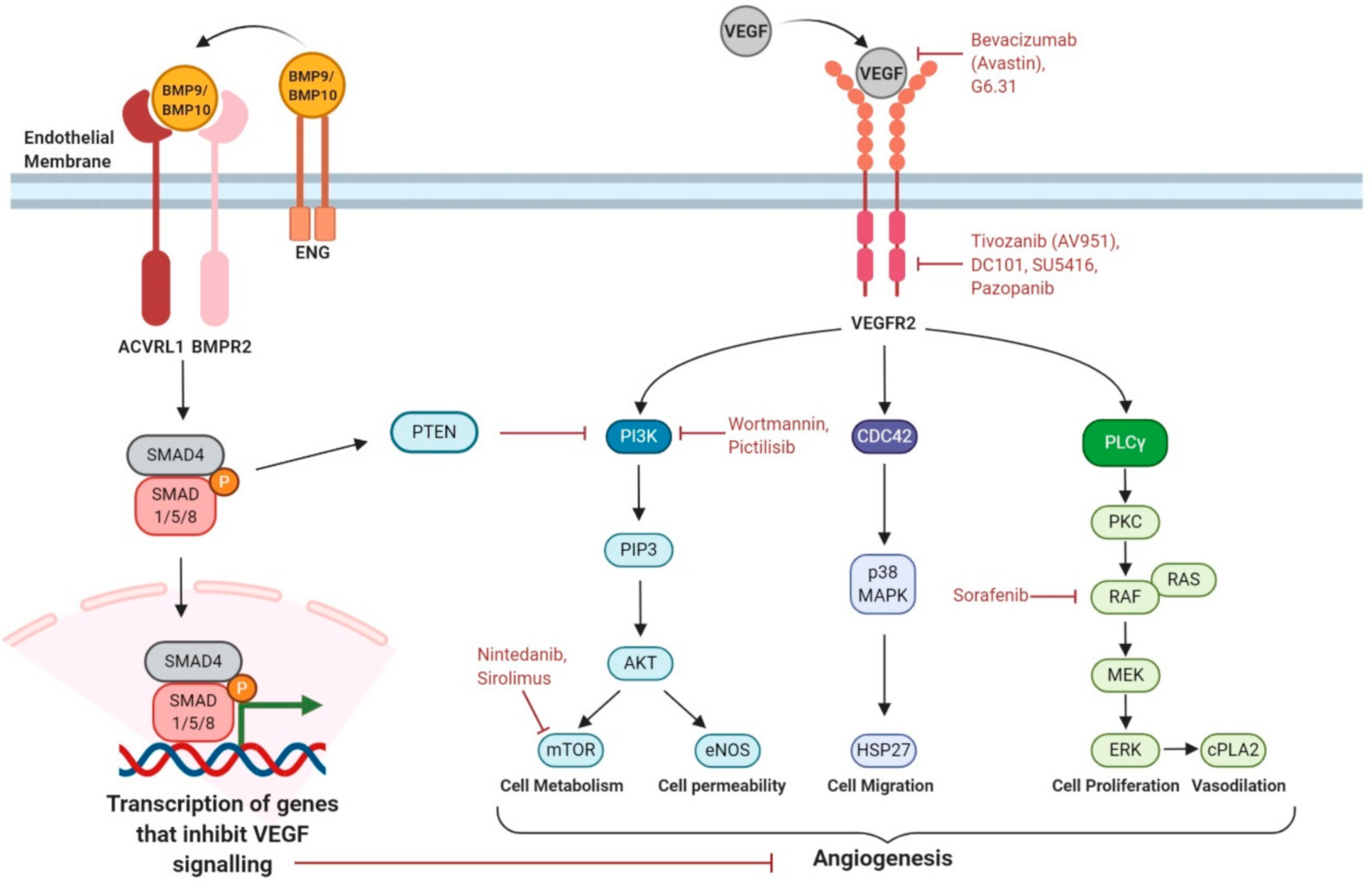
5.1. Increasing Expression of ENG or ACVRL1 Genes
5.2. Increasing BMP9/10 Ligand Availability
5.3. Targeting Proangiogenic Growth Factor Signalling
5.4. β-Blockers
5.5. Enhancing Pericyte–Endothelial Cell Interactions
6. Summary and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- McDonald, J.; Wooderchak-Donahue, W.; VanSant Webb, C.; Whitehead, K.; Stevenson, D.A.; Bayrak-Toydemir, P. Hereditary hemorrhagic telangiectasia: Genetics and molecular diagnostics in a new era. Front. Genet. 2015, 6, 1. [Google Scholar] [CrossRef] [PubMed]
- Gallione, C.J.; Repetto, G.M.; Legius, E.; Rustgi, A.K.; Schelley, S.L.; Tejpar, S.; Mitchell, G.; Drouin, E.; Westermann, C.J.; Marchuk, D.A. A combined syndrome of juvenile polyposis and hereditary haemorrhagic telangiectasia associated with mutations in MADH4 (SMAD4). Lancet 2004, 363, 852–859. [Google Scholar] [CrossRef]
- Wooderchak-Donahue, W.L.; McDonald, J.; O’Fallon, B.; Upton, P.D.; Li, W.; Roman, B.L.; Young, S.; Plant, P.; Fulop, G.T.; et al. BMP9 mutations cause a vascular-anomaly syndrome with phenotypic overlap with hereditary hemorrhagic telangiectasia. Am. J. Hum. Genet. 2013, 93, 530–537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bayrak-Toydemir, P.; McDonald, J.; Markewitz, B.; Lewin, S.; Miller, F.; Chou, L.S.; Gedge, F.; Tang, W.; Coon, H.; Mao, R. Genotype-phenotype correlation in hereditary hemorrhagic telangiectasia: Mutations and manifestations. Am. J. Med. Genet. A 2006, 140, 463–470. [Google Scholar] [CrossRef] [PubMed]
- Lesca, G.; Olivieri, C.; Burnichon, N.; Pagella, F.; Carette, M.F.; Gilbert-Dussardier, B.; Goizet, C.; Roume, J.; Rabilloud, M.; Saurin, J.C.; et al. Genotype-phenotype correlations in hereditary hemorrhagic telangiectasia: Data from the French-Italian HHT network. Genet. Med. 2007, 9, 14–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tual-Chalot, S.; Oh, S.P.; Arthur, H.M. Mouse models of hereditary hemorrhagic telangiectasia: Recent advances and future challenges. Front. Genet. 2015, 6, 25. [Google Scholar] [CrossRef] [Green Version]
- Bidart, M.; Ricard, N.; Levet, S.; Samson, M.; Mallet, C.; David, L.; Subileau, M.; Tillet, E.; Feige, J.J.; Bailly, S. BMP9 is produced by hepatocytes and circulates mainly in an active mature form complexed to its prodomain. Cell. Mol. Life Sci. 2012, 69, 313–324. [Google Scholar] [CrossRef]
- Chen, H.; Shi, S.; Acosta, L.; Li, W.; Lu, J.; Bao, S.; Chen, Z.; Yang, Z.; Schneider, M.D.; Chien, K.R.; et al. BMP10 is essential for maintaining cardiac growth during murine cardiogenesis. Development 2004, 131, 2219–2231. [Google Scholar] [CrossRef] [Green Version]
- Ricard, N.; Ciais, D.; Levet, S.; Subileau, M.; Mallet, C.; Zimmers, T.A.; Lee, S.J.; Bidart, M.; Feige, J.J.; Bailly, S. BMP9 and BMP10 are critical for postnatal retinal vascular remodeling. Blood 2012, 119, 6162–6171. [Google Scholar] [CrossRef] [Green Version]
- Salmon, R.M.; Guo, J.; Wood, J.H.; Tong, Z.; Beech, J.S.; Lawera, A.; Yu, M.; Grainger, D.J.; Reckless, J.; Morrell, N.W.; et al. Molecular basis of ALK1-mediated signalling by BMP9/BMP10 and their prodomain-bound forms. Nat. Commun. 2020, 11, 1621. [Google Scholar] [CrossRef] [Green Version]
- Castonguay, R.; Werner, E.D.; Matthews, R.G.; Presman, E.; Mulivor, A.W.; Solban, N.; Sako, D.; Pearsall, R.S.; Underwood, K.W.; Seehra, J.; et al. Soluble Endoglin Specifically Binds Bone Morphogenetic Proteins 9 and 10 via Its Orphan Domain, Inhibits Blood Vessel Formation, and Suppresses Tumor Growth. J. Biol. Chem. 2011, 286, 30034–30046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scharpfenecker, M.; van Dinther, M.; Liu, Z.; van Bezooijen, R.L.; Zhao, Q.; Pukac, L.; Lowik, C.W.; ten Dijke, P. BMP-9 signals via ALK1 and inhibits bFGF-induced endothelial cell proliferation and VEGF-stimulated angiogenesis. J. Cell Sci. 2007, 120, 964–972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- David, L.; Mallet, C.; Mazerbourg, S.; Feige, J.J.; Bailly, S. Identification of BMP9 and BMP10 as functional activators of the orphan activin receptor-like kinase 1 (ALK1) in endothelial cells. Blood 2007, 109, 1953–1961. [Google Scholar] [CrossRef] [PubMed]
- Saito, T.; Bokhove, M.; Croci, R.; Zamora-Caballero, S.; Han, L.; Letarte, M.; de Sanctis, D.; Jovine, L. Structural Basis of the Human Endoglin-BMP9 Interaction: Insights into BMP Signaling and HHT1. Cell Rep. 2017, 19, 1917–1928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lawera, A.; Tong, Z.; Thorikay, M.; Redgrave, R.E.; Cai, J.; van Dinther, M.; Morrell, N.W.; Afink, G.B.; Charnock-Jones, D.S.; Arthur, H.M.; et al. Role of soluble endoglin in BMP9 signaling. Proc. Natl. Acad. Sci. USA 2019, 116, 17800–17808. [Google Scholar] [CrossRef] [Green Version]
- Tillet, E.; Ouarne, M.; Desroches-Castan, A.; Mallet, C.; Subileau, M.; Didier, R.; Lioutsko, A.; Belthier, G.; Feige, J.J.; Bailly, S. A heterodimer formed by bone morphogenetic protein 9 (BMP9) and BMP10 provides most BMP biological activity in plasma. J. Biol. Chem. 2018, 293, 10963–10974. [Google Scholar] [CrossRef] [Green Version]
- Ola, R.; Dubrac, A.; Han, J.; Zhang, F.; Fang, J.S.; Larrivee, B.; Lee, M.; Urarte, A.A.; Kraehling, J.R.; Genet, G.; et al. PI3 kinase inhibition improves vascular malformations in mouse models of hereditary haemorrhagic telangiectasia. Nat. Commun. 2016, 7, 13650. [Google Scholar] [CrossRef]
- Ruiz, S.; Zhao, H.; Chandakkar, P.; Chatterjee, P.K.; Papoin, J.; Blanc, L.; Metz, C.N.; Campagne, F.; Marambaud, P. A mouse model of hereditary hemorrhagic telangiectasia generated by transmammary-delivered immunoblocking of BMP9 and BMP10. Sci. Rep. 2016, 5, 37366. [Google Scholar] [CrossRef]
- Laux, D.W.; Young, S.; Donovan, J.P.; Mansfield, C.J.; Upton, P.D.; Roman, B.L. Circulating Bmp10 acts through endothelial Alk1 to mediate flow-dependent arterial quiescence. Development 2013, 140, 3403–3412. [Google Scholar] [CrossRef] [Green Version]
- Capasso, T.L.; Li, B.; Volek, H.J.; Khalid, W.; Rochon, E.R.; Anbalagan, A.; Herdman, C.; Yost, H.J.; Villanueva, F.S.; Kim, K.; et al. BMP10-mediated ALK1 signaling is continuously required for vascular development and maintenance. Angiogenesis 2020, 23, 203–220. [Google Scholar] [CrossRef]
- Levet, S.; Ciais, D.; Merdzhanova, G.; Mallet, C.; Zimmers, T.A.; Lee, S.J.; Navarro, F.P.; Texier, I.; Feige, J.J.; Bailly, S.; et al. Bone morphogenetic protein 9 (BMP9) controls lymphatic vessel maturation and valve formation. Blood 2013, 122, 598–607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.; Brady Ridgway, J.; Sai, T.; Lai, J.; Warming, S.; Chen, H.; Roose-Girma, M.; Zhang, G.; Shou, W.; Yan, M. Context-dependent signaling defines roles of BMP9 and BMP10 in embryonic and postnatal development. Proc. Natl. Acad. Sci. USA 2013, 110, 11887–11892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Townson, S.A.; Martinez-Hackert, E.; Greppi, C.; Lowden, P.; Sako, D.; Liu, J.; Ucran, J.A.; Liharska, K.; Underwood, K.W.; Seehra, J.; et al. Specificity and structure of a high affinity activin receptor-like kinase 1 (ALK1) signaling complex. J. Biol. Chem. 2012, 287, 27313–27325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crist, A.M.; Lee, A.R.; Patel, N.R.; Westhoff, D.E.; Meadows, S.M. Vascular deficiency of Smad4 causes arteriovenous malformations: A mouse model of Hereditary Hemorrhagic Telangiectasia. Angiogenesis 2018, 21, 363–380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benn, A.; Alonso, F.; Mangelschots, J.; Genot, E.; Lox, M.; Zwijsen, A. BMP-SMAD1/5 Signaling Regulates Retinal Vascular Development. Biomolecules 2020, 10, 488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dupuis-Girod, S.; Bailly, S.; Plauchu, H. Hereditary hemorrhagic telangiectasia: From molecular biology to patient care. J. Thromb. Haemost. 2010, 8, 1447–1456. [Google Scholar] [CrossRef]
- Shovlin, C.L. Hereditary haemorrhagic telangiectasia: Pathophysiology, diagnosis and treatment. Blood Rev. 2010, 24, 203–219. [Google Scholar] [CrossRef] [Green Version]
- Snellings, D.A.; Gallione, C.J.; Clark, D.S.; Vozoris, N.T.; Faughnan, M.E.; Marchuk, D.A. Somatic Mutations in Vascular Malformations of Hereditary Hemorrhagic Telangiectasia Result in Bi-allelic Loss of ENG or ACVRL1. Am. J. Hum. Genet. 2019, 105, 894–906. [Google Scholar] [CrossRef]
- Li, C.; Guo, B.; Ding, S.; Rius, C.; Langa, C.; Kumar, P.; Bernabeu, C.; Kumar, S. TNF α down-regulates CD105 expression in vascular endothelial cells: A comparative study with TGF β 1. Anticancer Res. 2003, 23, 1189–1196. [Google Scholar]
- Fernandez, L.A.; Sanz-Rodriguez, F.; Zarrabeitia, R.; Perez-Molino, A.; Morales, C.; Restrepo, C.M.; Ramirez, J.R.; Coto, E.; Lenato, G.M.; Bernabeu, C.; et al. Mutation study of Spanish patients with hereditary hemorrhagic telangiectasia and expression analysis of Endoglin and ALK1. Hum. Mutat. 2006, 27, 295. [Google Scholar] [CrossRef]
- Kawasaki, K.; Freimuth, J.; Meyer, D.S.; Lee, M.M.; Tochimoto-Okamoto, A.; Benzinou, M.; Clermont, F.F.; Wu, G.; Roy, R.; Letteboer, T.G.; et al. Genetic variants of Adam17 differentially regulate TGFbeta signaling to modify vascular pathology in mice and humans. Proc. Natl. Acad. Sci. USA 2014, 111, 7723–7728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, S.O.; Wankhede, M.; Lee, Y.J.; Choi, E.J.; Fliess, N.; Choe, S.W.; Oh, S.H.; Walter, G.; Raizada, M.K.; Sorg, B.S.; et al. Real-time imaging of de novo arteriovenous malformation in a mouse model of hereditary hemorrhagic telangiectasia. J. Clin. Investig. 2009, 119, 3487–3496. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.W.; Harmon, C.; O’Farrelly, C. Liver immunology and its role in inflammation and homeostasis. Cell Mol. Immunol. 2016, 13, 267–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baeyens, N.; Larrivee, B.; Ola, R.; Hayward-Piatkowskyi, B.; Dubrac, A.; Huang, B.; Ross, T.D.; Coon, B.G.; Min, E.; Tsarfati, M.; et al. Defective fluid shear stress mechanotransduction mediates hereditary hemorrhagic telangiectasia. J. Cell Biol. 2016, 214, 807–816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corti, P.; Young, S.; Chen, C.Y.; Patrick, M.J.; Rochon, E.R.; Pekkan, K.; Roman, B.L. Interaction between alk1 and blood flow in the development of arteriovenous malformations. Development 2011, 138, 1573–1582. [Google Scholar] [CrossRef] [Green Version]
- Rochon, E.R.; Menon, P.G.; Roman, B.L. Alk1 controls arterial endothelial cell migration in lumenized vessels. Development 2016, 143, 2593–2602. [Google Scholar] [CrossRef] [Green Version]
- Jin, Y.; Muhl, L.; Burmakin, M.; Wang, Y.; Duchez, A.C.; Betsholtz, C.; Arthur, H.M.; Jakobsson, L. Endoglin prevents vascular malformation by regulating flow-induced cell migration and specification through VEGFR2 signalling. Nat. Cell Biol. 2017, 19, 639–652. [Google Scholar] [CrossRef] [Green Version]
- Lebrin, F.; Srun, S.; Raymond, K.; Martin, S.; van den Brink, S.; Freitas, C.; Breant, C.; Mathivet, T.; Larrivee, B.; Thomas, J.L.; et al. Thalidomide stimulates vessel maturation and reduces epistaxis in individuals with hereditary hemorrhagic telangiectasia. Nat. Med. 2010, 16, 420–428. [Google Scholar] [CrossRef]
- Chen, W.; Guo, Y.; Walker, E.J.; Shen, F.; Jun, K.; Oh, S.P.; Degos, V.; Lawton, M.T.; Tihan, T.; Davalos, D.; et al. Reduced mural cell coverage and impaired vessel integrity after angiogenic stimulation in the Alk1-deficient brain. Arterioscler. Thrombosis Vasc. Biol. 2013, 33, 305–310. [Google Scholar] [CrossRef] [Green Version]
- Winkler, E.A.; Birk, H.; Burkhardt, J.K.; Chen, X.; Yue, J.K.; Guo, D.; Rutledge, W.C.; Lasker, G.F.; Partow, C.; Tihan, T.; et al. Reductions in brain pericytes are associated with arteriovenous malformation vascular instability. J. Neurosurg. 2018, 129, 1464–1474. [Google Scholar] [CrossRef] [Green Version]
- Nikolaev, S.I.; Vetiska, S.; Bonilla, X.; Boudreau, E.; Jauhiainen, S.; Rezai Jahromi, B.; Khyzha, N.; DiStefano, P.V.; Suutarinen, S.; Kiehl, T.R.; et al. Somatic activating KRAS mutations in arteriovenous malformations of the brain. N. Eng. J. Med. 2018, 378, 250–261. [Google Scholar] [CrossRef] [PubMed]
- Fish, J.E.; Flores Suarez, C.P.; Boudreau, E.; Herman, A.M.; Gutierrez, M.C.; Gustafson, D.; DiStefano, P.V.; Cui, M.; Chen, Z.; De Ruiz, K.B.; et al. Somatic Gain of KRAS Function in the Endothelium Is Sufficient to Cause Vascular Malformations That Require MEK but Not PI3K Signaling. Circ. Res. 2020, 127, 727–743. [Google Scholar] [CrossRef] [PubMed]
- Tual-Chalot, S.; Garcia-Collado, M.; Redgrave, R.E.; Singh, E.; Davison, B.; Park, C.; Lin, H.; Luli, S.; Jin, Y.; Wang, Y.; et al. Loss of Endothelial Endoglin Promotes High-Output Heart Failure Through Peripheral Arteriovenous Shunting Driven by VEGF Signaling. Circ. Res. 2020, 126, 243–257. [Google Scholar] [CrossRef] [PubMed]
- Alsina-Sanchis, E.; Garcia-Ibanez, Y.; Figueiredo, A.M.; Riera-Domingo, C.; Figueras, A.; Matias-Guiu, X.; Casanovas, O.; Botella, L.M.; Pujana, M.A.; Riera-Mestre, A.; et al. ALK1 Loss Results in Vascular Hyperplasia in Mice and Humans Through PI3K Activation. Arterioscler. Thrombosis Vasc. Biol. 2018, 38, 1216–1229. [Google Scholar] [CrossRef] [Green Version]
- Mahmoud, M.; Allinson, K.R.; Zhai, Z.; Oakenfull, R.; Ghandi, P.; Adams, R.H.; Fruttiger, M.; Arthur, H.M. Pathogenesis of arteriovenous malformations in the absence of endoglin. Circ. Res. 2010, 106, 1425–1433. [Google Scholar] [CrossRef] [Green Version]
- Tual-Chalot, S.; Mahmoud, M.; Allinson, K.R.; Redgrave, R.E.; Zhai, Z.; Oh, S.P.; Fruttiger, M.; Arthur, H.M. Endothelial depletion of Acvrl1 in mice leads to arteriovenous malformations associated with reduced endoglin expression. PLoS ONE 2014, 9, e98646. [Google Scholar] [CrossRef] [Green Version]
- Iriarte, A.; Figueras, A.; Cerda, P.; Mora, J.M.; Jucgla, A.; Penin, R.; Vinals, F.; Riera-Mestre, A. PI3K (Phosphatidylinositol 3-Kinase) Activation and Endothelial Cell Proliferation in Patients with Hemorrhagic Hereditary Telangiectasia Type 1. Cells 2019, 8, 971. [Google Scholar] [CrossRef] [Green Version]
- Jakobsson, L.; Arthur, H.M. Oncogenes in Brain Arteriovenous Malformations. Circ. Res. 2020, 127, 744–746. [Google Scholar] [CrossRef]
- Dupuis-Girod, S.; Ginon, I.; Saurin, J.C.; Marion, D.; Guillot, E.; Decullier, E.; Roux, A.; Carette, M.F.; Gilbert-Dussardier, B.; Hatron, P.Y.; et al. Bevacizumab in patients with hereditary hemorrhagic telangiectasia and severe hepatic vascular malformations and high cardiac output. JAMA 2012, 307, 948–955. [Google Scholar] [CrossRef] [Green Version]
- Al-Samkari, H.; Kasthuri, R.S.; Parambil, J.G.; Albitar, H.A.; Almodallal, Y.A.; Vazquez, C.; Serra, M.M.; Dupuis-Girod, S.; Wilsen, C.B.; McWilliams, J.P.; et al. An international, multicenter study of intravenous bevacizumab for bleeding in hereditary hemorrhagic telangiectasia: The InHIBIT-Bleed study. Haematologica 3324. [Google Scholar] [CrossRef]
- Dupuis-Girod, S.; Fargeton, A.E.; Grobost, V.; Riviere, S.; Beaudoin, M.; Decullier, E.; Bernard, L.; Breant, V.; Colombet, B.; Philouze, P.; et al. Efficacy and Safety of a 0.1% Tacrolimus Nasal Ointment as a Treatment for Epistaxis in Hereditary Hemorrhagic Telangiectasia: A Double-Blind, Randomized, Placebo-Controlled, Multicenter Trial. J. Clin. Med. 2020, 9, 1262. [Google Scholar] [CrossRef] [PubMed]
- Faughnan, M.E.; Gossage, J.R.; Chakinala, M.M.; Oh, S.P.; Kasthuri, R.; Hughes, C.C.W.; McWilliams, J.P.; Parambil, J.G.; Vozoris, N.; Donaldson, J.; et al. Pazopanib may reduce bleeding in hereditary hemorrhagic telangiectasia. Angiogenesis 2019, 22, 145–155. [Google Scholar] [CrossRef] [PubMed]
- Dupuis-Girod, S.; Pitiot, V.; Bergerot, C.; Fargeton, A.E.; Beaudoin, M.; Decullier, E.; Breant, V.; Colombet, B.; Philouze, P.; Faure, F.; et al. Efficacy of TIMOLOL nasal spray as a treatment for epistaxis in hereditary hemorrhagic telangiectasia. A double-blind, randomized, placebo-controlled trial. Sci Rep 2019, 9, 11986. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, S.; Zhao, H.; Chandakkar, P.; Papoin, J.; Choi, H.; Nomura-Kitabayashi, A.; Patel, R.; Gillen, M.; Diao, L.; Chatterjee, P.K.; et al. Correcting Smad1/5/8, mTOR, and VEGFR2 treats pathology in hereditary hemorrhagic telangiectasia models. J. Clin. Investig. 2020, 130, 942–957. [Google Scholar] [CrossRef] [Green Version]
- Han, C.; Choe, S.W.; Kim, Y.H.; Acharya, A.P.; Keselowsky, B.G.; Sorg, B.S.; Lee, Y.J.; Oh, S.P. VEGF neutralization can prevent and normalize arteriovenous malformations in an animal model for hereditary hemorrhagic telangiectasia 2. Angiogenesis 2014, 17, 823–830. [Google Scholar] [CrossRef]
- Kim, Y.H.; Kim, M.J.; Choe, S.W.; Sprecher, D.; Lee, Y.J.; Oh, S.P. Selective effects of oral antiangiogenic tyrosine kinase inhibitors on an animal model of hereditary hemorrhagic telangiectasia. J. Thromb. Haemost. 2017, 15, 1095–1102. [Google Scholar] [CrossRef]
- Zhu, W.; Chen, W.; Zou, D.; Wang, L.; Bao, C.; Zhan, L.; Saw, D.; Wang, S.; Winkler, E.; Li, Z.; et al. Thalidomide Reduces Hemorrhage of Brain Arteriovenous Malformations in a Mouse Model. Stroke 2018, 49, 1232–1240. [Google Scholar] [CrossRef]
- Crist, A.M.; Zhou, X.; Garai, J.; Lee, A.R.; Thoele, J.; Ullmer, C.; Klein, C.; Zabaleta, J.; Meadows, S.M. Angiopoietin-2 Inhibition Rescues Arteriovenous Malformation in a Smad4 Hereditary Hemorrhagic Telangiectasia Mouse Model. Circulation 2019, 139, 2049–2063. [Google Scholar] [CrossRef]
- Ruiz, S.; Chandakkar, P.; Zhao, H.; Papoin, J.; Chatterjee, P.K.; Christen, E.; Metz, C.N.; Blanc, L.; Campagne, F.; Marambaud, P. Tacrolimus rescues the signaling and gene expression signature of endothelial ALK1 loss-of-function and improves HHT vascular pathology. Hum. Mol. Genet. 2017, 26, 4786–4798. [Google Scholar] [CrossRef]
- Albinana, V.; Sanz-Rodriguez, F.; Recio-Poveda, L.; Bernabeu, C.; Botella, L.M. Immunosuppressor FK506 increases endoglin and activin receptor-like kinase 1 expression and modulates transforming growth factor-beta1 signaling in endothelial cells. Mol. Pharmacol. 2011, 79, 833–843. [Google Scholar] [CrossRef] [Green Version]
- Spiekerkoetter, E.; Sung, Y.K.; Sudheendra, D.; Bill, M.; Aldred, M.A.; van de Veerdonk, M.C.; Vonk Noordegraaf, A.; Long-Boyle, J.; Dash, R.; Yang, P.C.; et al. Low-Dose FK506 (Tacrolimus) in End-Stage Pulmonary Arterial Hypertension. Am. J. Respir. Crit. Care Med. 2015, 192, 254–257. [Google Scholar] [CrossRef] [Green Version]
- Sommer, N.; Droege, F.; Gamen, K.E.; Geisthoff, U.; Gall, H.; Tello, K.; Richter, M.J.; Deubner, L.M.; Schmiedel, R.; Hecker, M.; et al. Treatment with low-dose tacrolimus inhibits bleeding complications in a patient with hereditary hemorrhagic telangiectasia and pulmonary arterial hypertension. Pulm. Circ. 2019, 9, 2045894018805406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hwan Kim, Y.; Vu, P.N.; Choe, S.W.; Jeon, C.J.; Arthur, H.M.; Vary, C.P.H.; Lee, Y.J.; Oh, S.P. Overexpression of Activin Receptor-Like Kinase 1 in Endothelial Cells Suppresses Development of Arteriovenous Malformations in Mouse Models of Hereditary Hemorrhagic Telangiectasia. Circ. Res. 2020, 127, 1122–1137. [Google Scholar] [CrossRef] [PubMed]
- Echevarria, L.; Aupy, P.; Goyenvalle, A. Exon-skipping advances for Duchenne muscular dystrophy. Hum. Mol. Genet. 2018, 27, R163–R172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keeling, K.M.; Xue, X.; Gunn, G.; Bedwell, D.M. Therapeutics based on stop codon readthrough. Annu. Rev. Genom. Hum. Genet. 2014, 15, 371–394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flieger, D.; Hainke, S.; Fischbach, W. Dramatic improvement in hereditary hemorrhagic telangiectasia after treatment with the vascular endothelial growth factor (VEGF) antagonist bevacizumab. Ann. Hematol. 2006, 85, 631–632. [Google Scholar] [CrossRef] [PubMed]
- David, L.; Mallet, C.; Keramidas, M.; Lamande, N.; Gasc, J.M.; Dupuis-Girod, S.; Plauchu, H.; Feige, J.J.; Bailly, S. Bone morphogenetic protein-9 is a circulating vascular quiescence factor. Circ. Res. 2008, 102, 914–922. [Google Scholar] [CrossRef]
- Sadick, H.; Riedel, F.; Naim, R.; Goessler, U.; Hormann, K.; Hafner, M.; Lux, A. Patients with hereditary hemorrhagic telangiectasia have increased plasma levels of vascular endothelial growth factor and transforming growth factor-beta1 as well as high ALK1 tissue expression. Haematologica 2005, 90, 818–828. [Google Scholar]
- Shao, E.S.; Lin, L.; Yao, Y.; Bostrom, K.I. Expression of vascular endothelial growth factor is coordinately regulated by the activin-like kinase receptors 1 and 5 in endothelial cells. Blood 2009, 114, 2197–2206. [Google Scholar] [CrossRef] [Green Version]
- Esposito, A.; Viale, G.; Curigliano, G. Safety, Tolerability, and Management of Toxic Effects of Phosphatidylinositol 3-Kinase Inhibitor Treatment in Patients With Cancer: A Review. JAMA Oncol. 2019, 5, 1347–1354. [Google Scholar] [CrossRef]
- Tian, H.; Huang, J.J.; Golzio, C.; Gao, X.; Hector-Greene, M.; Katsanis, N.; Blobe, G.C. Endoglin interacts with VEGFR2 to promote angiogenesis. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2018, 32, 2934–2949. [Google Scholar] [CrossRef] [Green Version]
- Thalgott, J.H.; Dos-Santos-Luis, D.; Hosman, A.E.; Martin, S.; Lamandé, N.; Bracquart, D.; Srun, S.; Galaris, G.; de Boer, H.C.; Tual-Chalot, S.; et al. Decreased expression of vascular endothelial growth factor receptor 1 contributes to the pathogenesis of hereditary hemorrhagic telangiectasia type 2. Circulation 2018, 138, 2698–2712. [Google Scholar] [CrossRef]
- Mei-Zahav, M.; Gendler, Y.; Bruckheimer, E.; Prais, D.; Birk, E.; Watad, M.; Goldschmidt, N.; Soudry, E. Topical Propranolol Improves Epistaxis Control in Hereditary Hemorrhagic Telangiectasia (HHT): A Randomized Double-Blind Placebo-Controlled Trial. J. Clin. Med. 2020, 9, 3130. [Google Scholar] [CrossRef]
- Harrison, L.; Kundra, A.; Jervis, P. The use of thalidomide therapy for refractory epistaxis in hereditary haemorrhagic telangiectasia: Systematic review. J. Laryngol. Otol. 2018, 132, 866–871. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, R.L.; Jonker, L.; Goumans, M.J.; Larsson, J.; Bouwman, P.; Karlsson, S.; Dijke, P.T.; Arthur, H.M.; Mummery, C.L. Defective paracrine signalling by TGFbeta in yolk sac vasculature of endoglin mutant mice: A paradigm for hereditary haemorrhagic telangiectasia. Development 2004, 131, 6237–6247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ojeda-Fernandez, L.; Barrios, L.; Rodriguez-Barbero, A.; Recio-Poveda, L.; Bernabeu, C.; Botella, L.M. Reduced plasma levels of Ang-2 and sEng as novel biomarkers in hereditary hemorrhagic telangiectasia (HHT). Clin. Chim. Acta 2010, 411, 494–499. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Drug | Target | Number of HHT Patients | Outcomes | Reference |
|---|---|---|---|---|
| Bevacizumab (Avastin) | Anti-VEGFA antibody | 24 | Reduced epistaxis and improved cardiac function in liver VM patients with HOHF | [49] |
| Bevacizumab (Avastin) | Anti-VEGFA antibody | 238 | Reduced epistaxis | [50] |
| Tacrolimus (FK506) | Increased activation ACVRL1 | 24 (+24 placebo) | Reduced epistaxis | [51] |
| Pazopanib | TKI | 7 | Some improvement in Hb and epistaxis | [52] |
| Timolol | β-adrenergic blocking agent | 28 (+28 placebo) | No change in epistaxis | [53] |
| Thalidomide | Increased PDGFB expression | 7 | Reduced epistaxis | [38] |
| Drug | Target | Mouse Model | Outcomes | Reference |
| Wortmannin, Pictilisib | PI3K inhibitor | Acvrl1-iKOe and Eng-iKOe neonates | Reduced retinal AVMs | [17,37] |
| Nintedanib and Sirolimus | TKI and mTOR inhibitor | Neonatal antibody blockade of BMP9/10 | Combination therapy reduced and reversed retinal AVMs | [54] |
| DC101 | Anti-VEGFR2 antibody | Eng-iKOe adult | Prevents AVMs and HOHF | [43] |
| G6.31 | Anti-VEGFA antibody | Acvrl1-iKOe adult | Prevention of wound induced dermal AVMs; possible reversal of established wound AVMs | [55] |
| Sorafenib and Pazopanib analogue (GW771806) | TKI | Acvrl1-iKOe adult | Each drug alone significantly improved Hb and GI bleeding but did not prevent wound-induced skin AVMs. | [56] |
| SU5416 | VEGFR2 inhibitor | Eng-iKOe neonate | Significant reduction in retinal AVM size | [37] |
| Thalidomide | Increased PDGFB expression | Eng+/− and Acvrl1-iKO adult | Increased SM coverage of dermal and cerebral vessels, reduced cerebral haemorrhage | [38,57] |
| LC10 | ANGPT2 inhibitor | Smad4-iKOe neonate | Prevents retinal AVMs | [58] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Snodgrass, R.O.; Chico, T.J.A.; Arthur, H.M. Hereditary Haemorrhagic Telangiectasia, an Inherited Vascular Disorder in Need of Improved Evidence-Based Pharmaceutical Interventions. Genes 2021, 12, 174. https://doi.org/10.3390/genes12020174
Snodgrass RO, Chico TJA, Arthur HM. Hereditary Haemorrhagic Telangiectasia, an Inherited Vascular Disorder in Need of Improved Evidence-Based Pharmaceutical Interventions. Genes. 2021; 12(2):174. https://doi.org/10.3390/genes12020174
Chicago/Turabian StyleSnodgrass, Ryan O., Timothy J. A. Chico, and Helen M. Arthur. 2021. "Hereditary Haemorrhagic Telangiectasia, an Inherited Vascular Disorder in Need of Improved Evidence-Based Pharmaceutical Interventions" Genes 12, no. 2: 174. https://doi.org/10.3390/genes12020174