
Should Patients with Kearns-Sayre Syndrome and Corneal Endothelial Failure Be Genotyped for a TCF4 Trinucleotide Repeat, Commonly Associated with Fuchs Endothelial Corneal Dystrophy?

{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
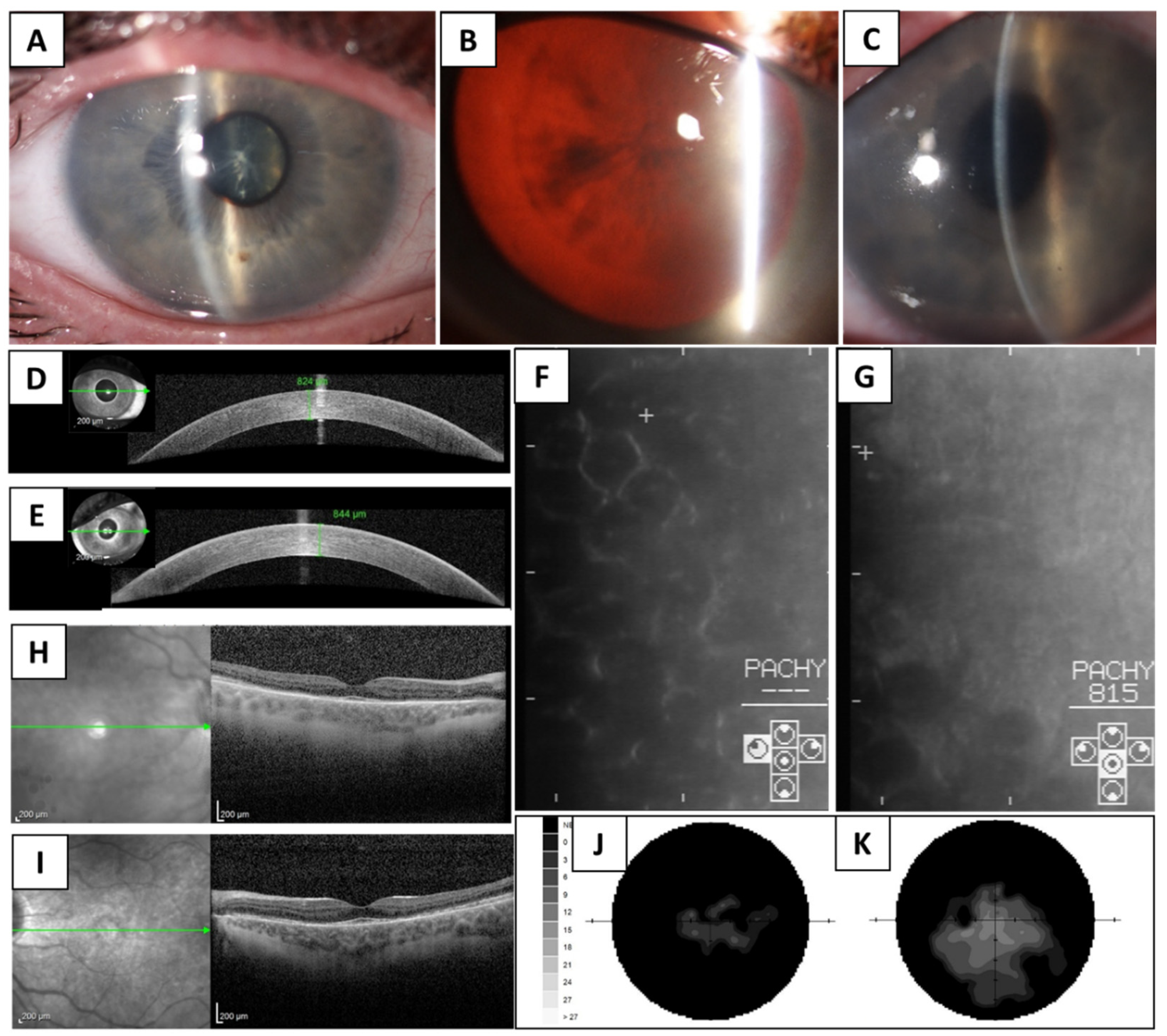
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Rowland, L.P. Molecular genetics, pseudogenetics, and clinical neurology. The Robert Wartenberg Lecture. Neurology 1983, 33, 1179–1195. [Google Scholar] [CrossRef] [PubMed]
- Mancuso, M.; Orsucci, D.; Angelini, C.; Bertini, E.; Carelli, V.; Comi, G.P.; Donati, M.A.; Federico, A.; Minetti, C.; Moggio, M.; et al. Redefining phenotypes associated with mitochondrial DNA single deletion. J. Neurol. 2015, 262, 1301–1309. [Google Scholar] [CrossRef] [PubMed]
- Finsterer, J.; Zarrouk-Mahjoub, S. Corneal Involvement in Kearns-Sayre Syndrome Responsive to Coenzyme-Q? Cornea 2016, 35, e39. [Google Scholar] [CrossRef] [PubMed]
- Ortiz, A.; Arias, J.; Cardenas, P.; Villamil, J.; Peralta, M.; Escaf, L.C.; Ortiz, J. Macular findings in Spectral Domain Optical Coherence Tomography and OCT Angiography in a patient with Kearns-Sayre syndrome. Int. J. Retin. Vitr. 2017, 3, 24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.; Medsinge, A.; Chauhan, B.; Wiest, C.; Scanga, H.; Monaghan, R.; Moore, W.H.; Nischal, K.K. Coenzyme Q10 in the Treatment of Corneal Edema in Kearns-Sayre: Is There an Application in Fuchs Endothelial Corneal Dystrophy? Cornea 2016, 35, 1250–1254. [Google Scholar] [CrossRef] [PubMed]
- Gonnermann, J.; Klamann, M.K.; Maier, A.K.; Bertelmann, E.; Schroeter, J.; von Au, K.; Joussen, A.M.; Torun, N. Descemet membrane endothelial keratoplasty in a child with corneal endothelial dysfunction in Kearns-Sayre syndrome. Cornea 2014, 33, 1232–1234. [Google Scholar] [CrossRef] [PubMed]
- Evans, C.J.; Liskova, P.; Dudakova, L.; Hrabcikova, P.; Horinek, A.; Jirsova, K.; Filipec, M.; Hardcastle, A.J.; Davidson, A.E.; Tuft, S.J. Identification of six novel mutations in ZEB1 and description of the associated phenotypes in patients with posterior polymorphous corneal dystrophy 3. Ann. Hum. Genet. 2015, 79, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Fautsch, M.P.; Wieben, E.D.; Baratz, K.H.; Bhattacharyya, N.; Sadan, A.N.; Hafford-Tear, N.J.; Tuft, S.J.; Davidson, A.E. TCF4-mediated Fuchs endothelial corneal dystrophy: Insights into a common trinucleotide repeat-associated disease. Prog. Retin. Eye Res. 2021, 81, 100883. [Google Scholar] [CrossRef] [PubMed]
- Bene, J.; Nadasi, E.; Kosztolanyi, G.; Mehes, K.; Melegh, B. Congenital cataract as the first symptom of a neuromuscular disease caused by a novel single large-scale mitochondrial DNA deletion. Eur. J. Hum. Genet. 2003, 11, 375–379. [Google Scholar] [CrossRef] [Green Version]
- Anteneova, N.; Kelifova, S.; Kolarova, H.; Vondrackova, A.; Tothova, I.; Liskova, P.; Magner, M.; Zamecnik, J.; Hansikova, H.; Zeman, J.; et al. The Phenotypic Spectrum of 47 Czech Patients with Single, Large-Scale Mitochondrial DNA Deletions. Brain Sci. 2020, 10, 766. [Google Scholar] [CrossRef]
- Dudakova, L.; Evans, C.J.; Pontikos, N.; Hafford-Tear, N.J.; Malinka, F.; Skalicka, P.; Horinek, A.; Munier, F.L.; Voide, N.; Studeny, P.; et al. The utility of massively parallel sequencing for posterior polymorphous corneal dystrophy type 3 molecular diagnosis. Exp. Eye Res. 2019, 182, 160–166. [Google Scholar] [CrossRef] [PubMed]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alfoldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef] [PubMed]
- Martin, A.R.; Williams, E.; Foulger, R.E.; Leigh, S.; Daugherty, L.C.; Niblock, O.; Leong, I.U.S.; Smith, K.R.; Gerasimenko, O.; Haraldsdottir, E.; et al. PanelApp crowdsources expert knowledge to establish consensus diagnostic gene panels. Nat. Genet. 2019, 51, 1560–1565. [Google Scholar] [CrossRef]
- Liskova, P.; Dudakova, L.; Evans, C.J.; Rojas Lopez, K.E.; Pontikos, N.; Athanasiou, D.; Jama, H.; Sach, J.; Skalicka, P.; Stranecky, V.; et al. Ectopic GRHL2 Expression Due to Non-coding Mutations Promotes Cell State Transition and Causes Posterior Polymorphous Corneal Dystrophy 4. Am. J. Hum. Genet. 2018, 102, 447–459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davidson, A.E.; Liskova, P.; Evans, C.J.; Dudakova, L.; Noskova, L.; Pontikos, N.; Hartmannova, H.; Hodanova, K.; Stranecky, V.; Kozmik, Z.; et al. Autosomal-Dominant Corneal Endothelial Dystrophies CHED1 and PPCD1 Are Allelic Disorders Caused by Non-coding Mutations in the Promoter of OVOL2. Am. J. Hum. Genet. 2016, 98, 75–89. [Google Scholar] [CrossRef] [Green Version]
- Wieben, E.D.; Aleff, R.A.; Tosakulwong, N.; Butz, M.L.; Highsmith, W.E.; Edwards, A.O.; Baratz, K.H. A common trinucleotide repeat expansion within the transcription factor 4 (TCF4, E2-2) gene predicts Fuchs corneal dystrophy. PLoS ONE 2012, 7, e49083. [Google Scholar] [CrossRef] [PubMed]
- Gilani, F.; Cortese, M.; Ambrosio, R.R., Jr.; Lopes, B.; Ramos, I.; Harvey, E.M.; Belin, M.W. Comprehensive anterior segment normal values generated by rotating Scheimpflug tomography. J. Cataract Refract. Surg. 2013, 39, 1707–1712. [Google Scholar] [CrossRef]
- Zarouchlioti, C.; Sanchez-Pintado, B.; Hafford Tear, N.J.; Klein, P.; Liskova, P.; Dulla, K.; Semo, M.; Vugler, A.A.; Muthusamy, K.; Dudakova, L.; et al. Antisense Therapy for a Common Corneal Dystrophy Ameliorates TCF4 Repeat Expansion-Mediated Toxicity. Am. J. Hum. Genet. 2018, 102, 528–539. [Google Scholar] [CrossRef] [Green Version]
- Bourne, W.M. Biology of the corneal endothelium in health and disease. Eye 2003, 17, 912–918. [Google Scholar] [CrossRef]
- Chang, T.S.; Johns, D.R.; Stark, W.J.; Drachman, D.B.; Green, W.R. Corneal decompensation in mitochondrial ophthalmoplegia plus (Kearns-Sayre) syndrome. A clinicopathologic case report. Cornea 1994, 13, 269–273. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dudakova, L.; Skalicka, P.; Davidson, A.E.; Sadan, A.N.; Chylova, M.; Jahnova, H.; Anteneova, N.; Tesarova, M.; Honzik, T.; Liskova, P. Should Patients with Kearns-Sayre Syndrome and Corneal Endothelial Failure Be Genotyped for a TCF4 Trinucleotide Repeat, Commonly Associated with Fuchs Endothelial Corneal Dystrophy? Genes 2021, 12, 1918. https://doi.org/10.3390/genes12121918
Dudakova L, Skalicka P, Davidson AE, Sadan AN, Chylova M, Jahnova H, Anteneova N, Tesarova M, Honzik T, Liskova P. Should Patients with Kearns-Sayre Syndrome and Corneal Endothelial Failure Be Genotyped for a TCF4 Trinucleotide Repeat, Commonly Associated with Fuchs Endothelial Corneal Dystrophy? Genes. 2021; 12(12):1918. https://doi.org/10.3390/genes12121918
Chicago/Turabian StyleDudakova, Lubica, Pavlina Skalicka, Alice E. Davidson, Amanda N. Sadan, Monika Chylova, Helena Jahnova, Nicole Anteneova, Marketa Tesarova, Tomas Honzik, and Petra Liskova. 2021. "Should Patients with Kearns-Sayre Syndrome and Corneal Endothelial Failure Be Genotyped for a TCF4 Trinucleotide Repeat, Commonly Associated with Fuchs Endothelial Corneal Dystrophy?" Genes 12, no. 12: 1918. https://doi.org/10.3390/genes12121918