
In Silico Analysis of Pathogenic CRB1 Single Nucleotide Variants and Their Amenability to Base Editing as a Potential Lead for Therapeutic Intervention

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
3. Results
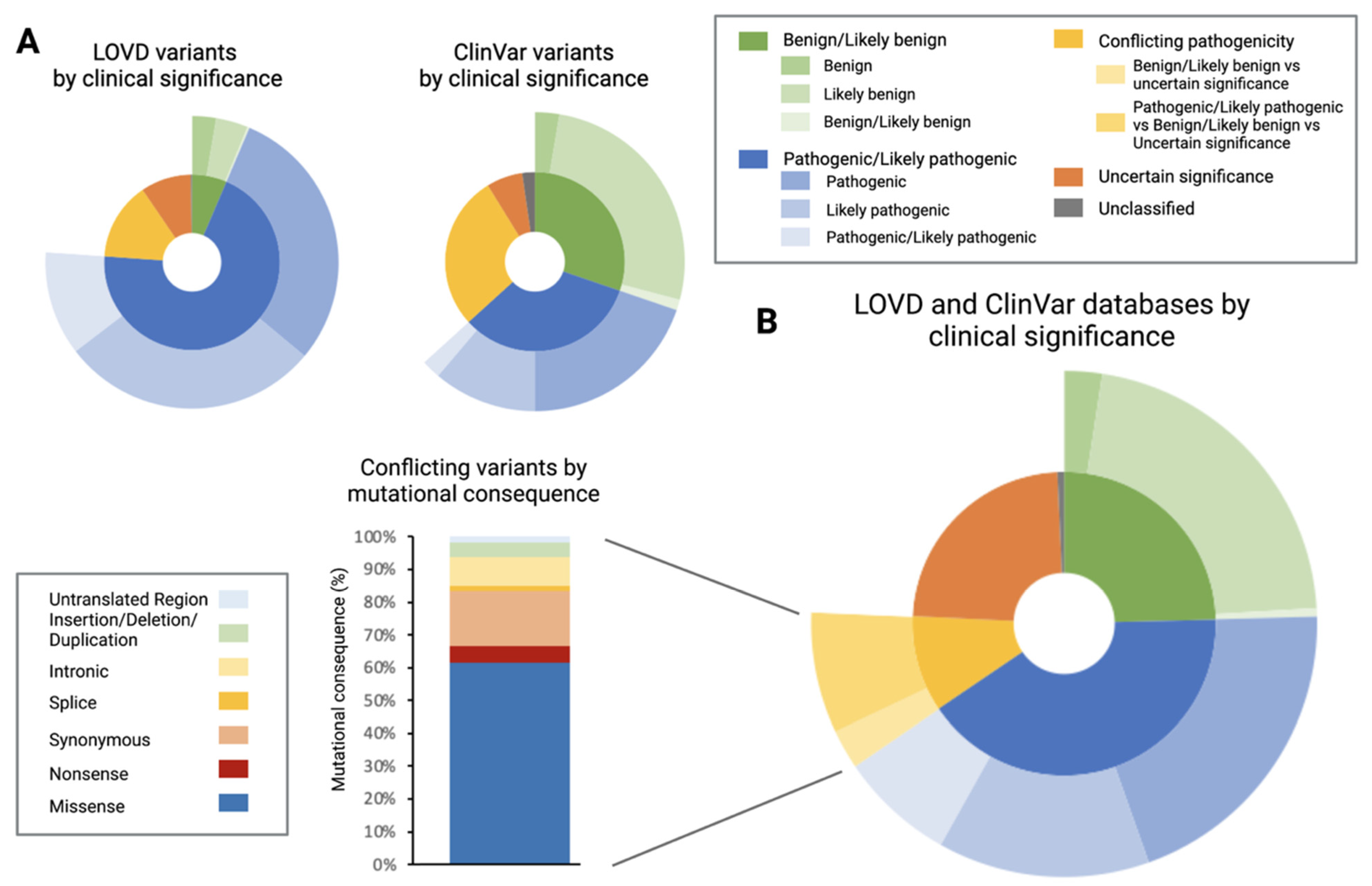
3.1. Characterisation of Leiden Open-Source Variation Database (LOVD) and ClinVar Database
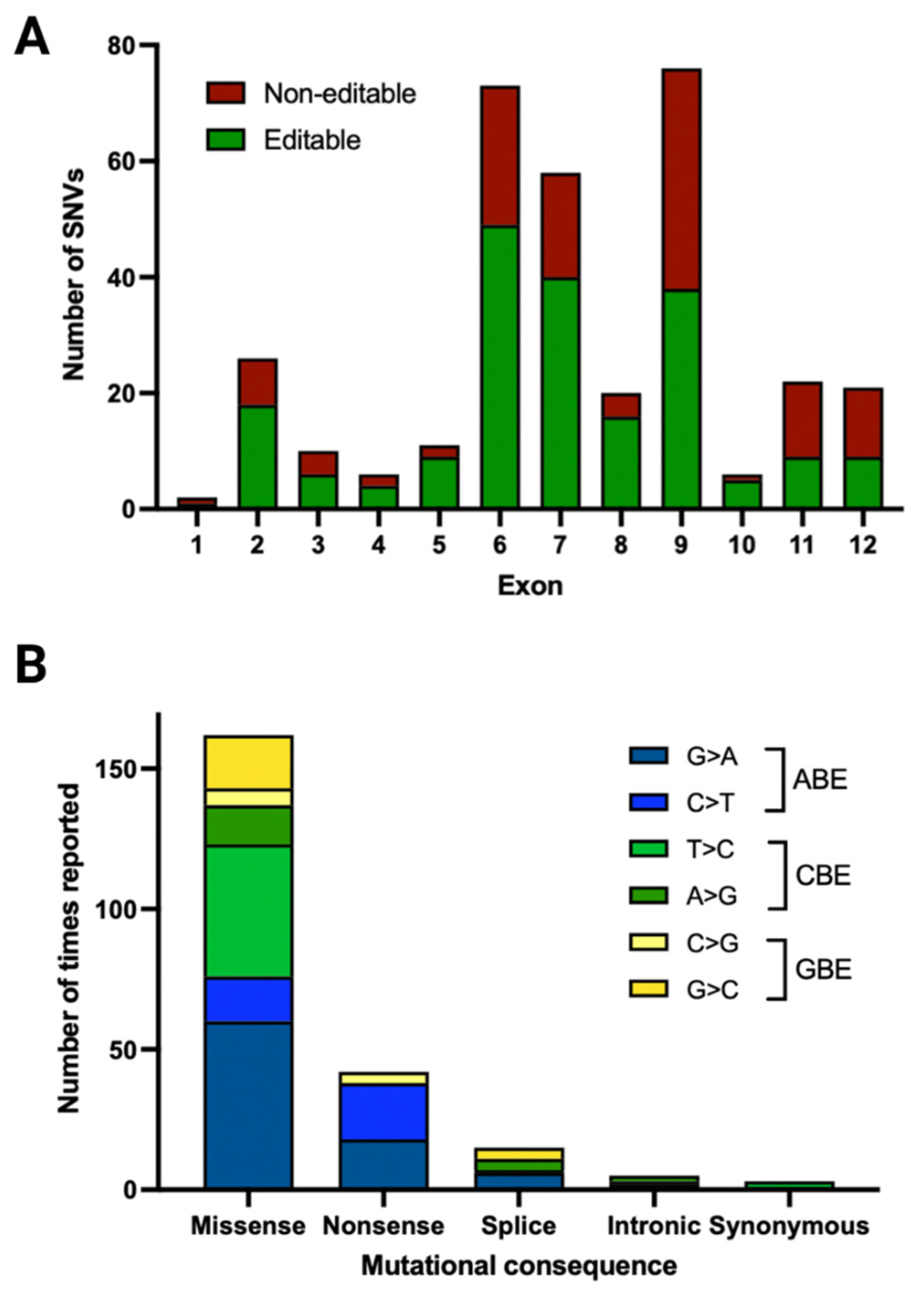
3.2. Pathogenic/Likely Pathogenic/Conflicting Pathogenic Single Nucleotide Variants (SNVs) by Mutational Consequence
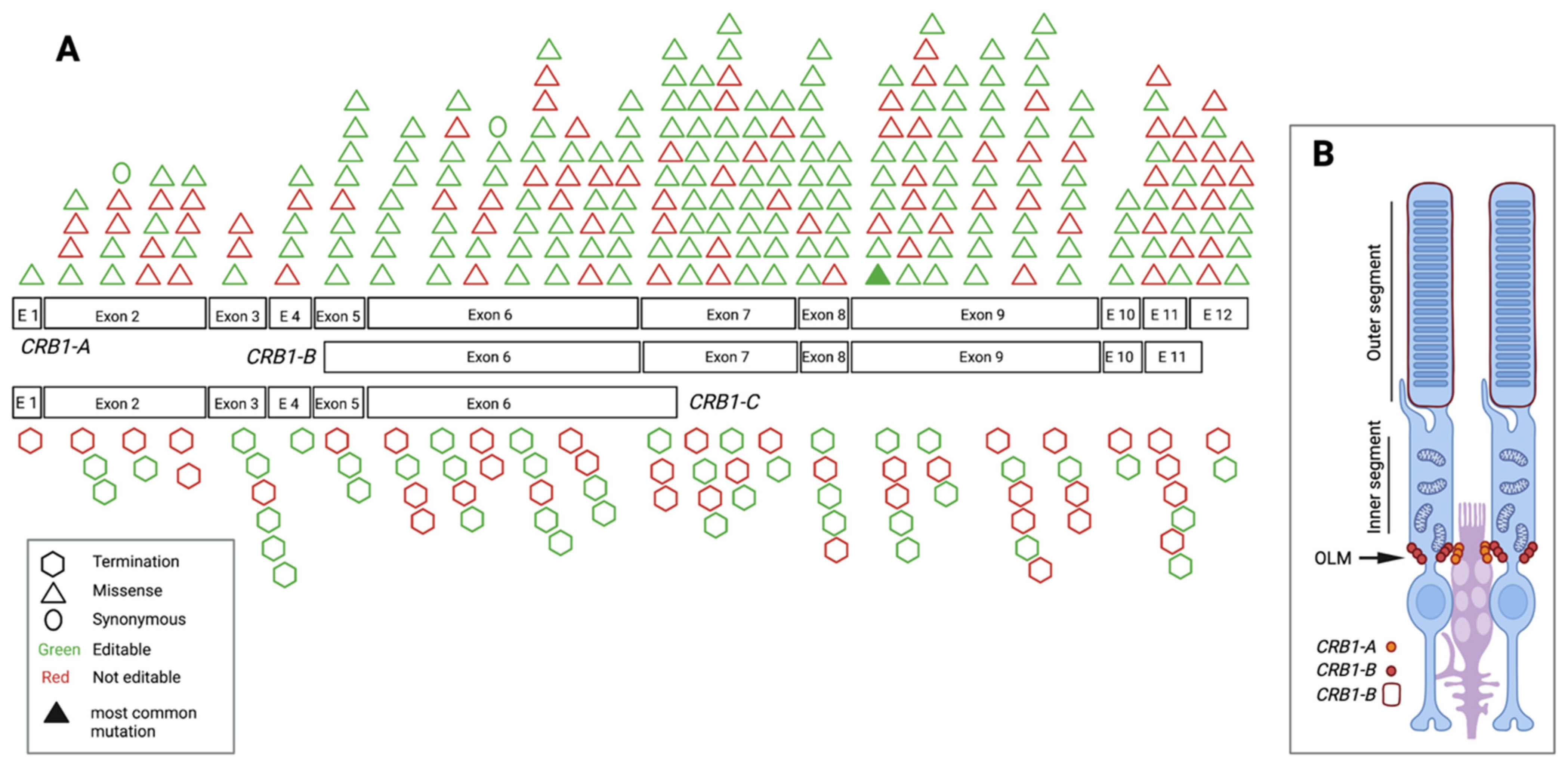
3.3. Pathogenic/Likely Pathogenic/Conflicting Pathogenic Exonic SNVs and Their Location
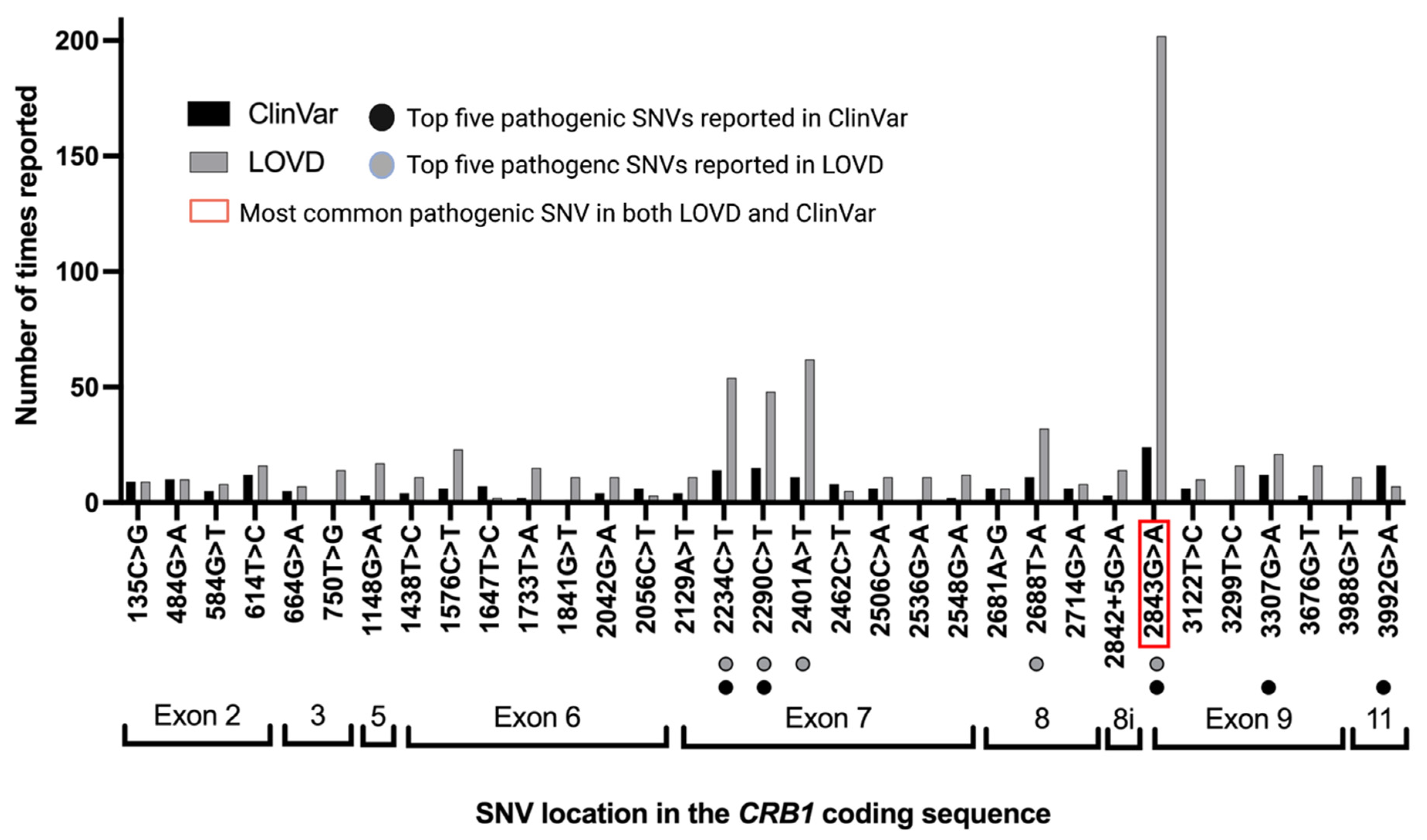
3.4. Pathogenic/Likely Pathogenic/Conflicting Pathogenic SNVs by Times Reported
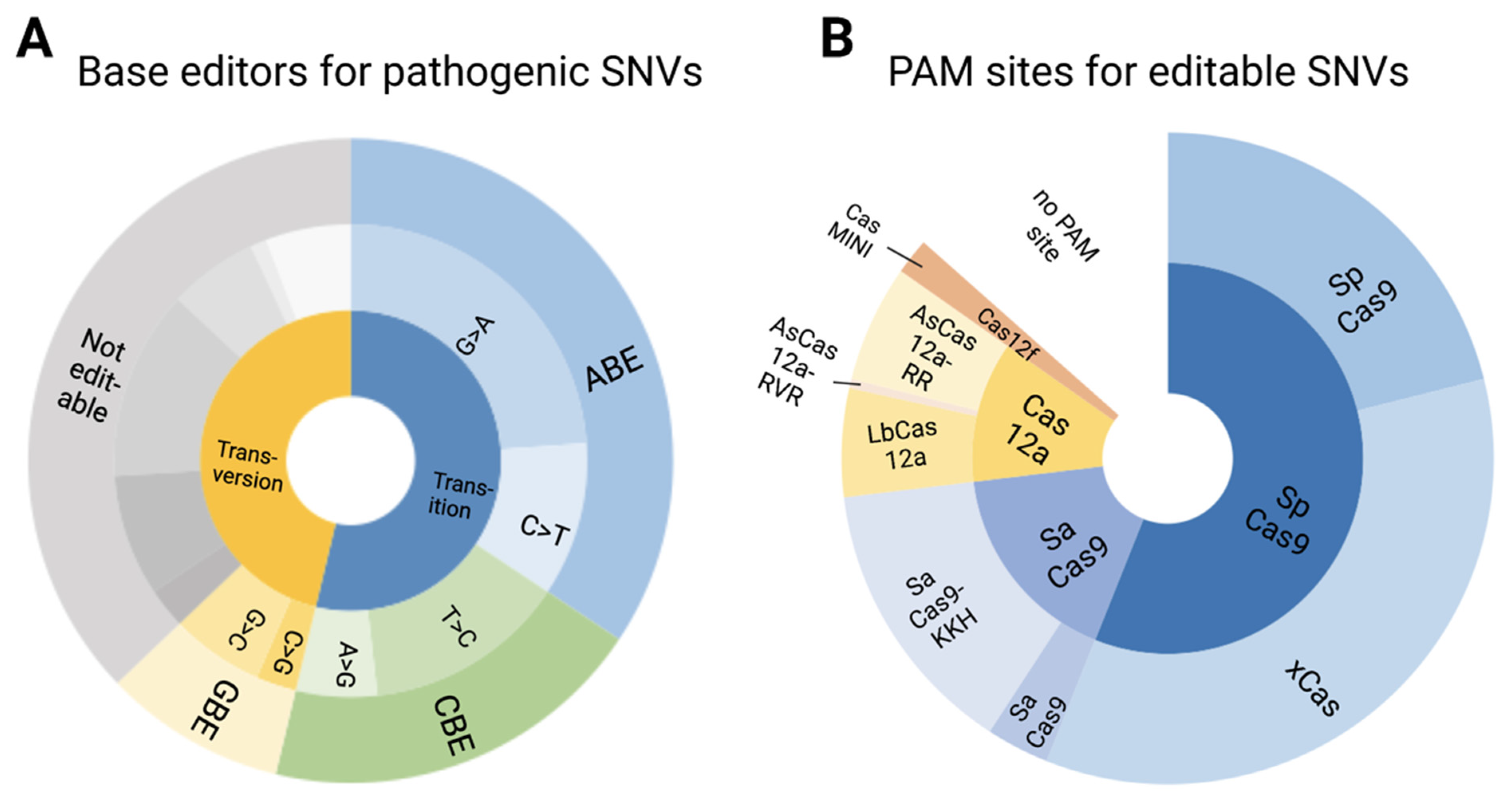
3.5. Pathogenic/Likely Pathogenic/Conflicting Pathogenic SNVs and Their Amenability to Base Editing
3.6. Editable Pathogenic SNVs and the Availability of PAM Sites
4. Discussion
4.1. Discrepancy between Databases
4.2. Mutational Consequences of Variants
4.3. Further Considerations
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A Programmable Dual-Rna-Guided DNA Endonuclease in Adaptive Bacterial Immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef] [PubMed]
- Komor, A.C.; Kim, Y.B.; Packer, M.S.; Zuris, J.A.; Liu, D.R. Programmable Editing of a Target Base in Genomic DNA without Double-Stranded DNA Cleavage. Nature 2016, 533, 420–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewis, C.A.; Crayle, J., Jr.; Zhou, S.; Swanstrom, R.; Wolfenden, R. Cytosine Deamination and the Precipitous Decline of Spontaneous Mutation During Earth’s History. Proc. Natl. Acad. Sci. USA 2016, 113, 8194–8199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaudelli, N.M.; Komor, A.C.; Rees, H.A.; Packer, M.S.; Badran, A.H.; Bryson, D.I.; Liu, D.R. Programmable Base Editing of a*T to G*C in Genomic DNA without DNA Cleavage. Nature 2017, 551, 464–471. [Google Scholar] [CrossRef]
- Gaudelli, N.M.; Lam, D.K.; Rees, H.A.; Sola-Esteves, N.M.; Barrera, L.A.; Born, D.A.; Edwards, A.; Gehrke, J.M.; Lee, S.J.; Liquori, A.J.; et al. Directed Evolution of Adenine Base Editors with Increased Activity and Therapeutic Application. Nat. Biotechnol. 2020, 38, 892–900. [Google Scholar] [CrossRef]
- Richter, M.F.; Zhao, K.T.; Eton, E.; Lapinaite, A.; Newby, G.A.; Thuronyi, B.; Wilson, C.; Koblan, L.W.; Zeng, J.; Bauer, D.E.; et al. Phage-Assisted Evolution of an Adenine Base Editor with Improved Cas Domain Compatibility and Activity. Nat. Biotechnol. 2020, 38, 883–891. [Google Scholar] [CrossRef]
- Komor, A.C.; Zhao, K.T.; Packer, M.S.; Gaudelli, N.M.; Waterbury, A.L.; Koblan, L.W.; Kim, Y.B.; Badran, A.H.; Liu, D.R. Improved Base Excision Repair Inhibition and Bacteriophage Mu Gam Protein Yields C:G-to-T:A Base Editors with Higher Efficiency and Product Purity. Sci. Adv. 2017, 3, eaao4774. [Google Scholar] [CrossRef] [Green Version]
- Yan, F.; Kuang, Y.; Ren, B.; Wang, J.; Zhang, D.; Lin, H.; Yang, B.; Zhou, X.; Zhou, H. Highly Efficient A.T to G.C Base Editing by Cas9n-Guided Trna Adenosine Deaminase in Rice. Mol. Plant 2018, 11, 631–634. [Google Scholar] [CrossRef] [Green Version]
- Kurt, I.C.; Zhou, R.; Iyer, S.; Garcia, S.P.; Miller, B.R.; Langner, L.M.; Grunewald, J.; Joung, J.K. Crispr C-to-G Base Editors for Inducing Targeted DNA Transversions in Human Cells. Nat. Biotechnol. 2021, 39, 41–46. [Google Scholar] [CrossRef]
- Zhao, D.; Li, J.; Li, S.; Xin, X.; Hu, M.; Price, M.A.; Rosser, S.J.; Bi, C.; Zhang, X. Glycosylase Base Editors Enable C-to-a and C-to-G Base Changes. Nat. Biotechnol. 2021, 39, 35–40. [Google Scholar] [CrossRef]
- Chen, L.; Park, J.E.; Paa, P.; Rajakumar, P.D.; Prekop, H.T.; Chew, Y.T.; Manivannan, S.N.; Chew, W.L. Programmable C:G to G:C Genome Editing with Crispr-Cas9-Directed Base Excision Repair Proteins. Nat. Commun. 2021, 12, 1384. [Google Scholar] [CrossRef]
- Kleinstiver, B.P.; Prew, M.S.; Tsai, S.Q.; Nguyen, N.T.; Topkar, V.V.; Zheng, Z.; Joung, J.K. Broadening the Targeting Range of Staphylococcus Aureus Crispr-Cas9 by Modifying Pam Recognition. Nat. Biotechnol. 2015, 33, 1293–1298. [Google Scholar] [CrossRef] [PubMed]
- Cehajic-Kapetanovic, J.; Xue, K.; de la Camara, C.M.; Nanda, A.; Davies, A.; Wood, L.J.; Salvetti, A.P.; Fischer, M.D.; Aylward, J.W.; Barnard, A.R.; et al. Initial Results from a First-in-Human Gene Therapy Trial on X-Linked Retinitis Pigmentosa Caused by Mutations in Rpgr. Nat. Med. 2020, 26, 354–359. [Google Scholar] [CrossRef]
- Jacobson, S.G.; Cideciyan, A.V.; Ratnakaram, R.; Heon, E.; Schwartz, S.B.; Roman, A.J.; Peden, M.C.; Aleman, T.S.; Boye, S.L.; Sumaroka, A.; et al. Gene Therapy for Leber Congenital Amaurosis Caused by Rpe65 Mutations: Safety and Efficacy in 15 Children and Adults Followed up to 3 Years. Arch. Ophthalmol. 2012, 130, 9–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bainbridge, J.W.; Mehat, M.S.; Sundaram, V.; Robbie, S.J.; Barker, S.E.; Ripamonti, C.; Georgiadis, A.; Mowat, F.M.; Beattie, S.G.; Gardner, P.J.; et al. Long-Term Effect of Gene Therapy on Leber’s Congenital Amaurosis. N. Engl. J. Med. 2015, 372, 1887–1897. [Google Scholar] [CrossRef] [Green Version]
- Hu, J.H.; Miller, S.M.; Geurts, M.H.; Tang, W.; Chen, L.; Sun, N.; Zeina, C.M.; Gao, X.; Rees, H.A.; Lin, Z.; et al. Evolved Cas9 Variants with Broad Pam Compatibility and High DNA Specificity. Nature 2018, 556, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Endo, M.; Mikami, M.; Endo, A.; Kaya, H.; Itoh, T.; Nishimasu, H.; Nureki, O.; Toki, S. Genome Editing in Plants by Engineered Crispr-Cas9 Recognizing Ng Pam. Nat. Plants 2019, 5, 14–17. [Google Scholar] [CrossRef]
- Kleinstiver, B.P.; Sousa, A.A.; Walton, R.T.; Tak, Y.E.; Hsu, J.Y.; Clement, K.; Welch, M.M.; Horng, J.E.; Malagon-Lopez, J.; Scarfo, I.; et al. Engineered Crispr-Cas12a Variants with Increased Activities and Improved Targeting Ranges for Gene, Epigenetic and Base Editing. Nat. Biotechnol. 2019, 37, 276–282. [Google Scholar] [CrossRef]
- Li, X.; Wang, Y.; Liu, Y.; Yang, B.; Wang, X.; Wei, J.; Lu, Z.; Zhang, Y.; Wu, J.; Huang, X.; et al. Base Editing with a Cpf1-Cytidine Deaminase Fusion. Nat. Biotechnol. 2018, 36, 324–327. [Google Scholar] [CrossRef]
- Xu, X.; Chemparathy, A.; Zeng, L.; Kempton, H.R.; Shang, S.; Nakamura, M.; Qi, L.S. Engineered Miniature Crispr-Cas System for Mammalian Genome Regulation and Editing. Mol. Cell 2021, 81, 4333–4345. [Google Scholar] [CrossRef]
- Den Hollander, A.I.; Brink, J.B.T.; de Kok, Y.J.; van Soest, S.; van den Born, L.I.; van Driel, M.A.; van de Pol, D.J.; Payne, A.M.; Bhattacharya, S.S.; Kellner, U.; et al. Mutations in a Human Homologue of Drosophila Crumbs Cause Retinitis Pigmentosa (Rp12). Nat. Genet. 1999, 23, 217–221. [Google Scholar] [CrossRef]
- Den Hollander, A.I.; Heckenlively, J.R.; van den Born, L.I.; de Kok, Y.J.; van der Velde-Visser, S.D.; Kellner, U.; Jurklies, B.; van Schooneveld, M.J.; Blankenagel, A.; Rohrschneider, K.; et al. Leber Congenital Amaurosis and Retinitis Pigmentosa with Coats-Like Exudative Vasculopathy Are Associated with Mutations in the Crumbs Homologue 1 (Crb1) Gene. Am. J. Hum. Genet. 2001, 69, 198–203. [Google Scholar] [CrossRef] [Green Version]
- Bujakowska, K.; Audo, I.; Mohand-Said, S.; Lancelot, M.E.; Antonio, A.; Germain, A.; Leveillard, T.; Letexier, M.; Saraiva, J.P.; Lonjou, C.; et al. Crb1 Mutations in Inherited Retinal Dystrophies. Hum. Mutat. 2012, 33, 306–315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corton, M.; Tatu, S.D.; Avila-Fernandez, A.; Vallespin, E.; Tapias, I.; Cantalapiedra, D.; Blanco-Kelly, F.; Riveiro-Alvarez, R.; Bernal, S.; Garcia-Sandoval, B.; et al. High Frequency of Crb1 Mutations as Cause of Early-Onset Retinal Dystrophies in the Spanish Population. Orphanet J. Rare Dis. 2013, 8, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vallespin, E.; Cantalapiedra, D.; Riveiro-Alvarez, R.; Wilke, R.; Aguirre-Lamban, J.; Avila-Fernandez, A.; Lopez-Martinez, M.A.; Gimenez, A.; Trujillo-Tiebas, M.J.; Ramos, C.; et al. Mutation Screening of 299 Spanish Families with Retinal Dystrophies by Leber Congenital Amaurosis Genotyping Microarray. Investig. Ophthalmol. Vis. Sci. 2007, 48, 5653–5661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tee, J.J.L.; Yang, Y.; Kalitzeos, A.; Webster, A.; Bainbridge, J.; Michaelides, M. Natural History Study of Retinal Structure, Progression, and Symmetry Using Ellipzoid Zone Metrics in Rpgr-Associated Retinopathy. Am. J. Ophthalmol. 2019, 198, 111–123. [Google Scholar] [CrossRef] [Green Version]
- Ray, T.A.; Cochran, K.; Kozlowski, C.; Wang, J.; Alexander, G.; Cady, M.A.; Spencer, W.J.; Ruzycki, P.A.; Clark, B.S.; Laeremans, A.; et al. Comprehensive Identification of Mrna Isoforms Reveals the Diversity of Neural Cell-Surface Molecules with Roles in Retinal Development and Disease. Nat. Commun. 2020, 11, 3328. [Google Scholar] [CrossRef]
- Bennett, J.; Wellman, J.; Marshall, K.A.; McCague, S.; Ashtari, M.; DiStefano-Pappas, J.; Elci, O.U.; Chung, D.C.; Sun, J.; Wright, J.F.; et al. Safety and Durability of Effect of Contralateral-Eye Administration of Aav2 Gene Therapy in Patients with Childhood-Onset Blindness Caused by Rpe65 Mutations: A Follow-on Phase 1 Trial. Lancet 2016, 388, 661–672. [Google Scholar] [CrossRef] [Green Version]
- Cideciyan, A.V.; Hauswirth, W.W.; Aleman, T.S.; Kaushal, S.; Schwartz, S.B.; Boye, S.L.; Windsor, E.A.; Conlon, T.J.; Sumaroka, A.; Pang, J.J.; et al. Human Rpe65 Gene Therapy for Leber Congenital Amaurosis: Persistence of Early Visual Improvements and Safety at 1 Year. Hum. Gene Ther. 2009, 20, 999–1004. [Google Scholar] [CrossRef] [Green Version]
- Buck, T.M.; Vos, R.M.; Alves, C.H.; Wijnholds, J. Aav-Crb2 Protects against Vision Loss in an Inducible Crb1 Retinitis Pigmentosa Mouse Model. Mol. Ther. Methods Clin. Dev. 2021, 20, 423–441. [Google Scholar] [CrossRef]
- Pellissier, L.P.; Quinn, P.M.; Alves, C.H.; Vos, R.M.; Klooster, J.; Flannery, J.G.; Heimel, J.A.; Wijnholds, J. Gene Therapy into Photoreceptors and Muller Glial Cells Restores Retinal Structure and Function in Crb1 Retinitis Pigmentosa Mouse Models. Hum. Mol. Genet. 2015, 24, 3104–3118. [Google Scholar] [CrossRef] [Green Version]
- Fry, L.E.; McClements, M.E.; MacLaren, R.E. Analysis of Pathogenic Variants Correctable with Crispr Base Editing among Patients with Recessive Inherited Retinal Degeneration. JAMA Ophthalmol. 2021, 139, 319–328. [Google Scholar] [CrossRef] [PubMed]
- Kantor, A.; McClements, M.E.; MacLaren, R.E. Crispr-Cas9 DNA Base-Editing and Prime-Editing. Int. J. Mol. Sci 2020, 21, 6240. [Google Scholar] [CrossRef] [PubMed]
- Koblan, L.W.; Doman, J.L.; Wilson, C.; Levy, J.M.; Tay, T.; Newby, G.A.; Maianti, J.P.; Raguram, A.; Liu, D.R. Improving Cytidine and Adenine Base Editors by Expression Optimization and Ancestral Reconstruction. Nat. Biotechnol. 2018, 36, 843–846. [Google Scholar] [CrossRef] [PubMed]
- Suh, S.; Choi, E.H.; Leinonen, H.; Foik, A.T.; Newby, G.A.; Yeh, W.H.; Dong, Z.; Kiser, P.D.; Lyon, D.C.; Liu, D.R.; et al. Restoration of Visual Function in Adult Mice with an Inherited Retinal Disease Via Adenine Base Editing. Nat. Biomed. Eng. 2021, 5, 169–178. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.B.; Komor, A.C.; Levy, J.M.; Packer, M.S.; Zhao, K.T.; Liu, D.R. Increasing the Genome-Targeting Scope and Precision of Base Editing with Engineered Cas9-Cytidine Deaminase Fusions. Nat. Biotechnol. 2017, 35, 371–376. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Zhang, X.; Wang, L.; Yin, S.; Zhu, B.; Xie, L.; Duan, Q.; Hu, H.; Zheng, R.; Wei, Y.; et al. Increasing Targeting Scope of Adenosine Base Editors in Mouse and Rat Embryos through Fusion of Tada Deaminase with Cas9 Variants. Protein Cell 2018, 9, 814–819. [Google Scholar] [CrossRef] [Green Version]
- Anzalone, A.V.; Randolph, P.B.; Davis, J.R.; Sousa, A.A.; Koblan, L.W.; Levy, J.M.; Chen, P.J.; Wilson, C.; Newby, G.A.; Raguram, A.; et al. Search-and-Replace Genome Editing without Double-Strand Breaks or Donor DNA. Nature 2019, 576, 149–157. [Google Scholar] [CrossRef]
- Fokkema, I.F.; Kroon, M.; Hernandez, J.A.L.; Asscheman, D.; Lugtenburg, I.; Hoogenboom, J.; den Dunnen, J.T. The Lovd3 Platform: Efficient Genome-Wide Sharing of Genetic Variants. Eur. J. Hum. Genet. 2021, 29, 1796–1803. [Google Scholar] [CrossRef]
- Li, Y.; Wang, H.; Peng, J.; Gibbs, R.A.; Lewis, R.A.; Lupski, J.R.; Mardon, G.; Chen, R. Mutation Survey of Known Lca Genes and Loci in the Saudi Arabian Population. Investig. Ophthalmol. Vis. Sci. 2009, 50, 1336–1343. [Google Scholar] [CrossRef]
- Lotery, A.J.; Jacobson, S.G.; Fishman, G.A.; Weleber, R.G.; Fulton, A.B.; Namperumalsamy, P.; Heon, E.; Levin, A.V.; Grover, S.; Rosenow, J.R.; et al. Mutations in the Crb1 Gene Cause Leber Congenital Amaurosis. Arch. Ophthalmol. 2001, 119, 415–420. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Krokan, H.E.; Drablos, F.; Slupphaug, G. Uracil in DNA—Occurrence, Consequences and Repair. Oncogene 2002, 21, 8935–8948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Z.; Yang, H.; Colosi, P. Effect of Genome Size on Aav Vector Packaging. Mol. Ther. 2010, 18, 80–86. [Google Scholar] [CrossRef]
- Carvalho, L.S.; Turunen, H.T.; Wassmer, S.J.; Luna-Velez, M.V.; Xiao, R.; Bennett, J.; Vandenberghe, L.H. Evaluating Efficiencies of Dual Aav Approaches for Retinal Targeting. Front. Neurosci. 2017, 11, 503. [Google Scholar] [CrossRef]
- Levy, J.M.; Yeh, W.H.; Pendse, N.; Davis, J.R.; Hennessey, E.; Butcher, R.; Koblan, L.W.; Comander, J.; Liu, Q.; Liu, D.R. Cytosine and Adenine Base Editing of the Brain, Liver, Retina, Heart and Skeletal Muscle of Mice Via Adeno-Associated Viruses. Nat. Biomed. Eng. 2020, 4, 97–110. [Google Scholar] [CrossRef]
- Trapani, I.; Puppo, A.; Auricchio, A. Vector Platforms for Gene Therapy of Inherited Retinopathies. Prog. Retin. Eye Res. 2014, 43, 108–128. [Google Scholar] [CrossRef] [Green Version]
- Gruter, O.; Kostic, C.; Crippa, S.V.; Perez, M.T.; Zografos, L.; Schorderet, D.F.; Munier, F.L.; Arsenijevic, Y. Lentiviral Vector-Mediated Gene Transfer in Adult Mouse Photoreceptors Is Impaired by the Presence of a Physical Barrier. Gene. Ther. 2005, 12, 942–947. [Google Scholar] [CrossRef] [Green Version]
- Stevanovic, M.; Piotter, E.; McClements, M.E.; MacLaren, R.E. Crispr Systems Suitable for Single Aav Vector Delivery. Curr. Gene Ther. 2021, 21. [Google Scholar] [CrossRef]
- Cremers, F.P.M.; Lee, W.; Collin, R.W.J.; Allikmets, R. Clinical Spectrum, Genetic Complexity and Therapeutic Approaches for Retinal Disease Caused by Abca4 Mutations. Prog. Retin. Eye Res. 2020, 79, 100861. [Google Scholar] [CrossRef]
- McCarty, N.S.; Graham, A.E.; Studena, L.; Ledesma-Amaro, R. Multiplexed Crispr Technologies for Gene Editing and Transcriptional Regulation. Nat. Commun. 2020, 11, 1281. [Google Scholar] [CrossRef]
- Xue, K.; Jolly, J.K.; Barnard, A.R.; Rudenko, A.; Salvetti, A.P.; Patricio, M.I.; Edwards, T.L.; Groppe, M.; Orlans, H.O.; Tolmachova, T.; et al. Beneficial Effects on Vision in Patients Undergoing Retinal Gene Therapy for Choroideremia. Nat. Med. 2018, 24, 1507–1512. [Google Scholar] [CrossRef] [PubMed]
- Reichel, F.F.; Dauletbekov, D.L.; Klein, R.; Peters, T.; Ochakovski, G.A.; Seitz, I.P.; Wilhelm, B.; Ueffing, M.; Biel, M.; Wissinger, B.; et al. Aav8 Can Induce Innate and Adaptive Immune Response in the Primate Eye. Mol. Ther. 2017, 25, 2648–2660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chandler, L.C.; McClements, M.E.; Yusuf, I.H.; de la Camara, C.M.; MacLaren, R.E.; Xue, K. Characterizing the Cellular Immune Response to Subretinal Aav Gene Therapy in the Murine Retina. Mol. Ther. Methods Clin. Dev. 2021, 22, 52–65. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cas Family | PAM Sequence 5’-3’ | PAM Location Relative to Target Base | Editing Window = | Previously Coupled with BE Class | Size (in aa) | ||
|---|---|---|---|---|---|---|---|
| Cas9 | SpCas9 | NGG | downstream | 4–8 | ABE7.10 [4], ABE8 [6], ABEmax [34] BE3 [2], BE4 [7], BE4max [34] GBE [9] | 1368 | |
| xCas/SpCas-NG* | NG | downstream | 4–8 | ABE7.10 [16,35] BE3 [16] GBE* [9] | |||
| SaCas9 | NNGRRT | downstream | 4–12 | ABE7.10, ABE8 [6] BE3 [36], BE4 [7] | 1053 | ||
| SaCas9-KKH | NNNRRT | downstream | 2–15 | ABE7.10 [37], ABE8 [6] BE3 [36] | |||
| Cas12 | Cas12a | LbCas12a | TTTV | upstream | 8–13 | ABE8 [6] BE3 [19] | 1228 |
| enAsCas12a-RR enAsCas12a-RVR | TATV, TYCV | upstream | 8–13 | ABE8 [6] BE3 [19] | |||
| Cas12f | CasMINI | TTTV | upstream | 3–4 | ABE8 [20] | 529 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bellingrath, J.-S.; McClements, M.E.; Kaukonen, M.; Fischer, M.D.; MacLaren, R.E. In Silico Analysis of Pathogenic CRB1 Single Nucleotide Variants and Their Amenability to Base Editing as a Potential Lead for Therapeutic Intervention. Genes 2021, 12, 1908. https://doi.org/10.3390/genes12121908
Bellingrath J-S, McClements ME, Kaukonen M, Fischer MD, MacLaren RE. In Silico Analysis of Pathogenic CRB1 Single Nucleotide Variants and Their Amenability to Base Editing as a Potential Lead for Therapeutic Intervention. Genes. 2021; 12(12):1908. https://doi.org/10.3390/genes12121908
Chicago/Turabian StyleBellingrath, Julia-Sophia, Michelle E. McClements, Maria Kaukonen, Manuel Dominik Fischer, and Robert E. MacLaren. 2021. "In Silico Analysis of Pathogenic CRB1 Single Nucleotide Variants and Their Amenability to Base Editing as a Potential Lead for Therapeutic Intervention" Genes 12, no. 12: 1908. https://doi.org/10.3390/genes12121908