
LAMA2 Nonsense Variant in an Italian Greyhound with Congenital Muscular Dystrophy

 , , , ,
, , , ,
Abstract
:1. Introduction
2. Materials and Methods
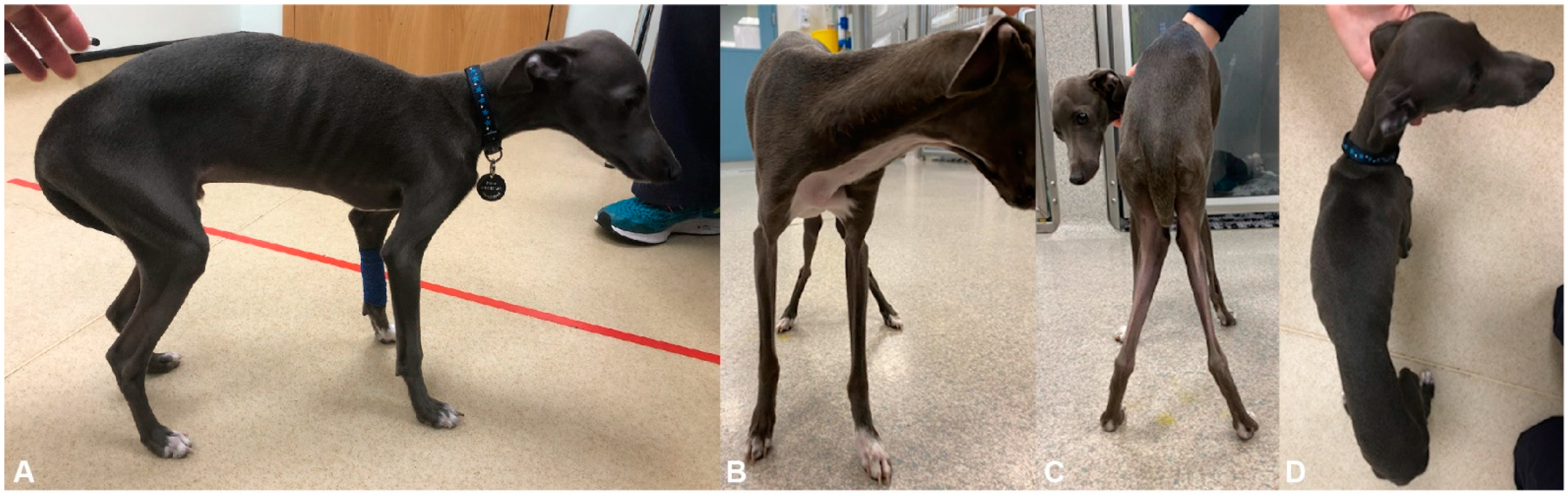
2.1. Clinical Examination
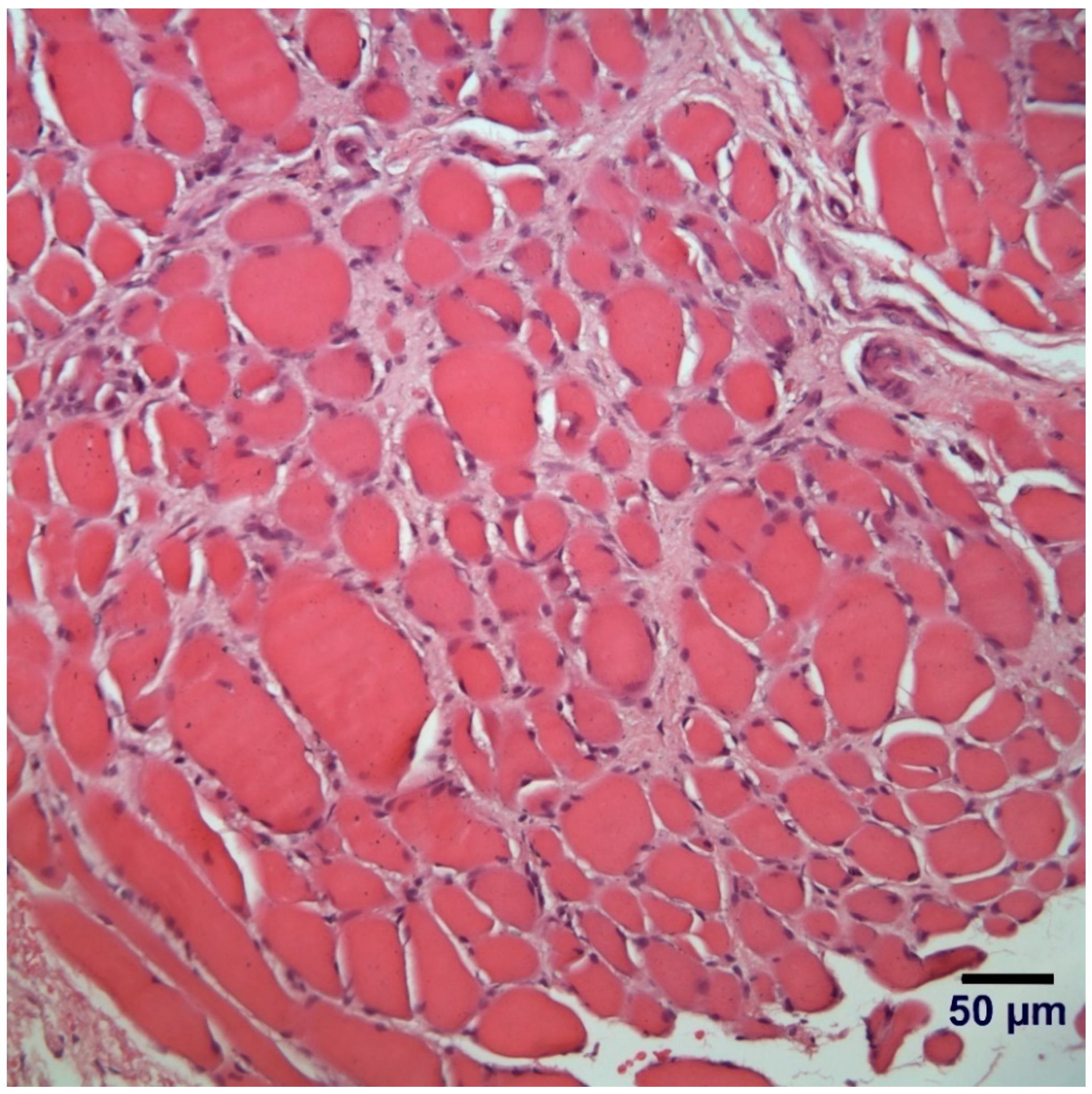
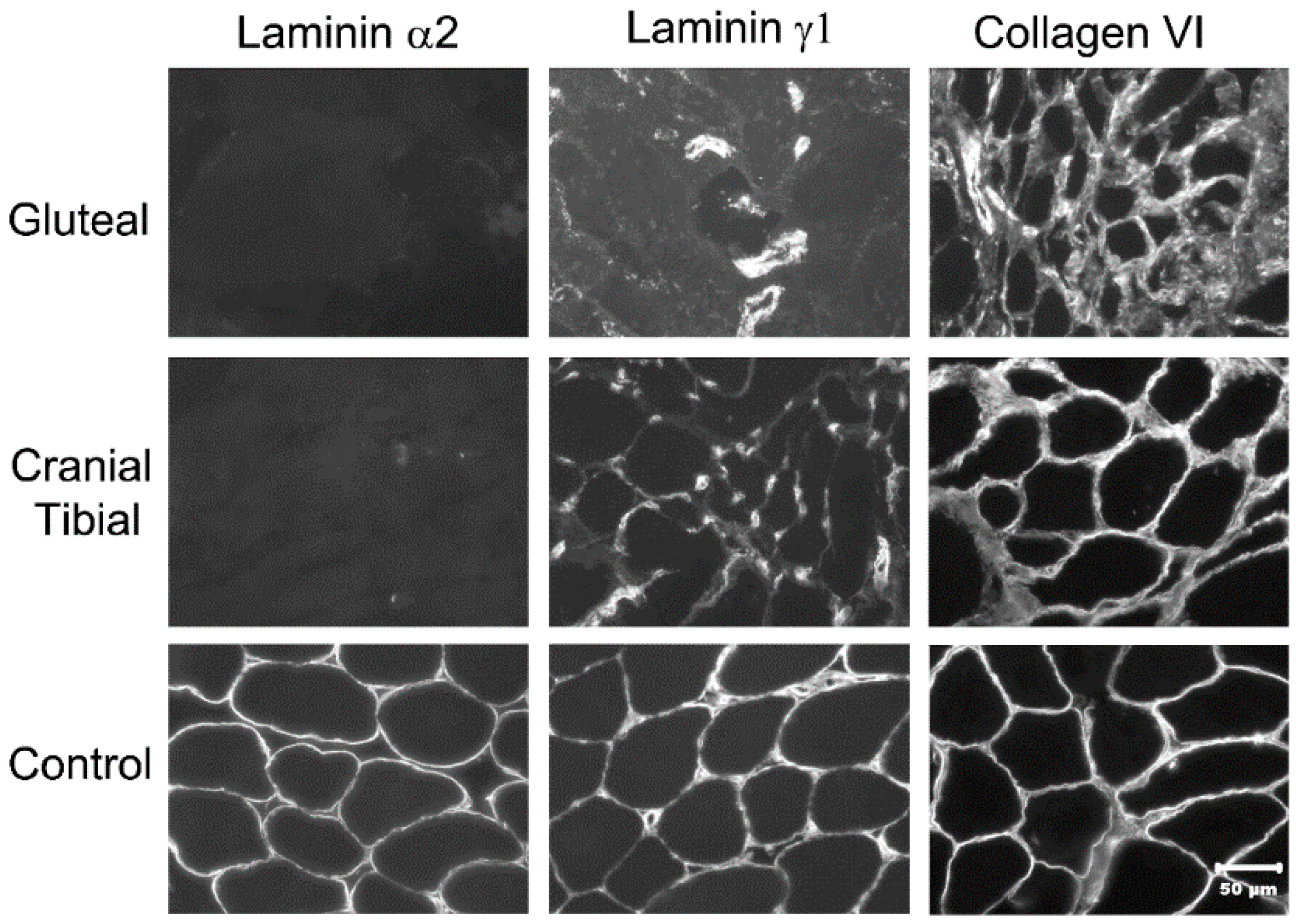
2.2. Histopathology and Immunofluorescent Labeling
2.3. Animals and DNA Extraction
2.4. Whole-Genome Sequencing
2.5. Variant Calling
2.6. Gene Analysis
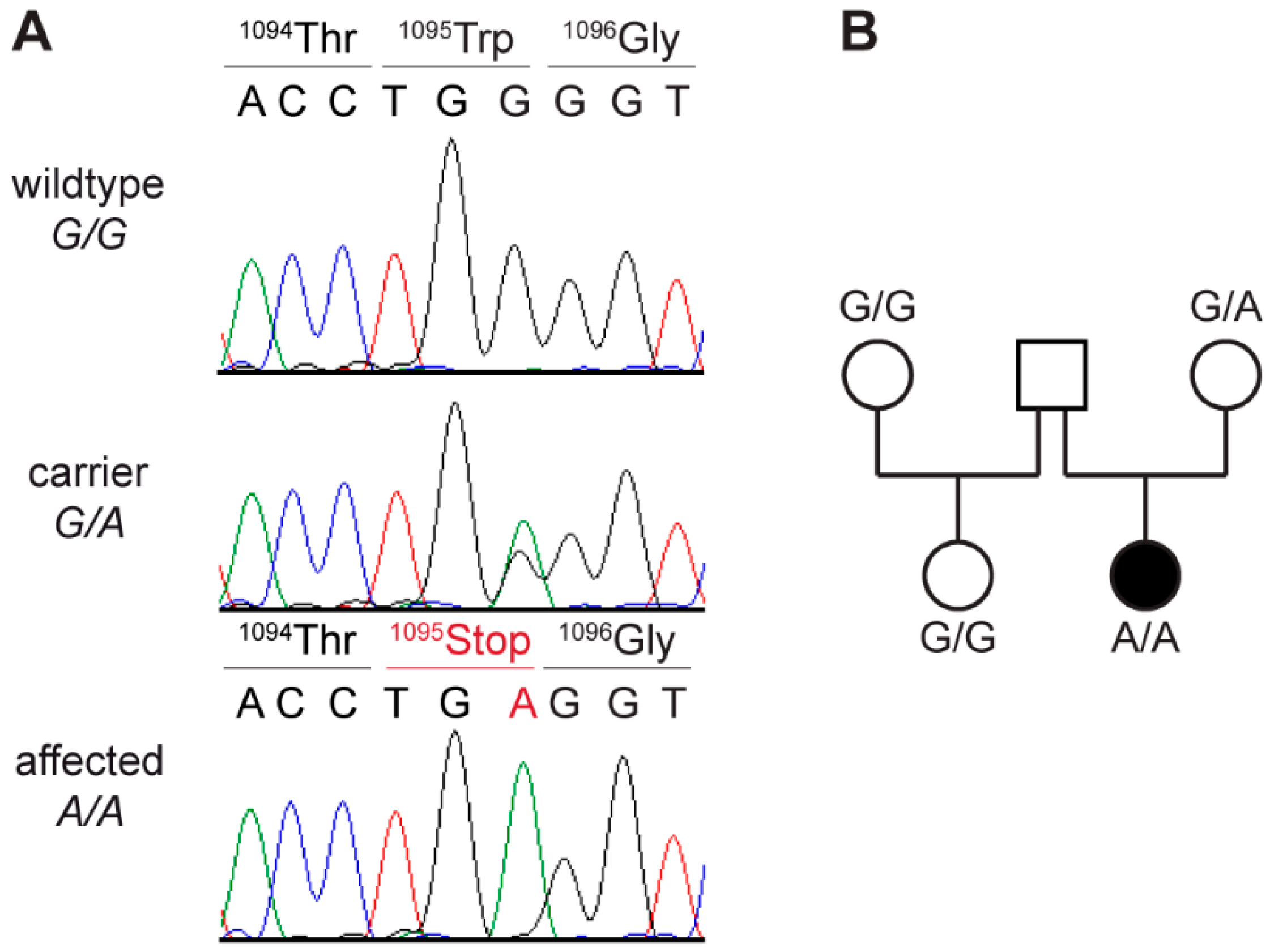
2.7. PCR and Sanger Sequencing
3. Results
3.1. Clinical History and Examination
3.2. Laboratory and Electrodiagnostic Examinations
3.3. Electrodiagnostic Testing
3.4. Clinical Outcome
3.5. Histopathological Examination
3.6. Genetic Analysis
3.7. Immunofluorescent Labeling of Laminin α2 in Muscle Cryosections
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zambon, A.A.; Muntoni, F. Congenital muscular dystrophies: What is new? Neuromuscul. Disord. 2021, 31, 931–942. [Google Scholar] [CrossRef] [PubMed]
- Butterfield, R.J. Congenital Muscular Dystrophy and Congenital Myopathy. Contin. Lifelong Learn. Neurol. 2019, 25, 1640–1661. [Google Scholar] [CrossRef] [PubMed]
- Bertini, E.; D’Amico, A.; Gualandi, F.; Petrini, S. Congenital Muscular Dystrophies: A Brief Review. Semin. Pediatr. Neurol. 2011, 18, 277–288. [Google Scholar] [CrossRef] [Green Version]
- Sframeli, M.; Sarkozy, A.; Bertoli, M.; Astrea, G.; Hudson, J.; Scoto, M.; Mein, R.; Yau, M.; Phadke, R.; Feng, L.; et al. Congenital muscular dystrophies in the UK population: Clinical and molecular spectrum of a large cohort diagnosed over a 12-year period. Neuromuscul. Disord. 2017, 27, 793–803. [Google Scholar] [CrossRef]
- Peat, R.A.; Smith, J.M.; Compton, A.G.; Baker, N.L.; Pace, R.A.; Burkin, D.J.; Kaufman, S.J.; Lamande, S.R.; North, K.N. Diagnosis and etiology of congenital muscular dystrophy. Neurology 2008, 71, 312–321. [Google Scholar] [CrossRef]
- Shelton, G.D. Muscular disorders. In Textbook of Veterinary Internal Medicine; Ettinger, S.J., Feldman, E.C., Côté, E., Eds.; Elsevier: St. Louis, MO, USA, 2017; pp. 5215–5228. ISBN 9780323312110. [Google Scholar]
- Steffen, F.; Bilzer, T.; Brands, J.; Golini, L.; Jagannathan, V.; Wiedmer, M.; Drögemüller, M.; Drögemüller, C.; Leeb, T. A nonsense variant in COL6A1 in Landseer Dogs with muscular dystrophy. G3 Genes Genomes Genet. 2015, 5, 2611–2617. [Google Scholar] [CrossRef] [Green Version]
- Bolduc, V.; Minor, K.M.; Hu, Y.; Kaur, R.; Friedenberg, S.G.; Van Buren, S.; Guo, L.T.; Glennon, J.C.; Marioni-Henry, K.; Mickelson, J.R.; et al. Pathogenic variants in COL6A3 cause Ullrich-like congenital muscular dystrophy in young Labrador Retriever dogs. Neuromuscul. Disord. 2020, 30, 360–367. [Google Scholar] [CrossRef]
- Shelton, G.D.; Minor, K.M.; Guo, L.T.; Friedenberg, S.G.; Cullen, J.N.; Hord, J.M.; Venzke, D.; Anderson, M.E.; Devereaux, M.; Prouty, S.J.; et al. Muscular dystrophy-dystroglycanopathy in a family of Labrador Retrievers with a LARGE 1 mutation. Neuromuscul. Disord. 2021, in press. [Google Scholar] [CrossRef]
- Shelton, G.D.; Minor, K.M.; Thomovsky, S.; Guo, L.T.; Friedenberg, S.G.; Cullen, J.N.; Mickelson, J.R. Congenital muscular dystrophy in a dog with a LAMA2 gene deletion. J. Vet. Intern. Med. 2021. [Google Scholar]
- Dubowitz, V.; Sewry, C.A.; Oldfors, A. Histological and histochemical stains and reactions. In Muscle Biopsy: A Practical Approach, 4th ed.; Dubowitz, V., Sewry, C.A., Oldfors, A., Eds.; Saunders Elsevier: Oxford, UK, 2013; pp. 16–27. [Google Scholar]
- Leivo, I.; Engvall, E. Merosin, a protein specific for basement membranes of Schwann cells, striated muscle, and trophoblast, is expressed late in nerve and muscle development. Proc. Natl. Acad. Sci. USA 1988, 85, 1544–1548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aumailley, M.; Brucknertuderman, L.; Carter, W.; Deutzmann, R.; Edgar, D.; Ekblom, P.; Engel, J.; Engvall, E.; Hohenester, E.; Jones, J. A simplified laminin nomenclature. Matrix Biol. 2005, 24, 326–332. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.T.; Moore, S.A.; Forcales, S.; Engvall, E.; Shelton, G.D. Evaluation of commercial dysferlin antibodies on canine, mouse and human skeletal muscle. Neuromuscul. Disord. 2010, 20, 820–825. [Google Scholar] [CrossRef]
- Jagannathan, V.; Drögemüller, C.; Leeb, T.; Dog Biomedical Variant Database Consortium (DBVDC). A comprehensive biomedical variant catalogue based on whole genome sequences of 582 dogs and eight wolves. Anim. Genet. 2019, 50, 695–704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [Green Version]
- Cingolani, P.; Platts, A.; Wang, L.L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef] [Green Version]
- Swallow, J.S.; Griffiths, I.R. Age related changes in the motor nerve conduction velocity in dogs. Res. Vet. Sci. 1977, 23, 29–32. [Google Scholar] [CrossRef]
- Sims, M.H.; Redding, R.W. Maturation of nerve conduction velocity and the evoked muscle potential in the dog. Am. J. Vet. Res. 1980, 41, 1247–1252. [Google Scholar]
- Brands, J.; Steffen, F.; Spennes, J.; Leeb, T.; Bilzer, T. COL6A1 related muscular dystrophy in Landseer dogs: A canine model for Ullrich congenital muscular dystrophy. Muscle Nerve 2021, 63, 608–616. [Google Scholar] [CrossRef]
- Shelton, G.D.; Liu, L.A.; Guo, L.T.; Smith, G.K.; Christiansen, J.S.; Thomas, W.B.; Smith, M.O.; Kline, K.L.; March, P.A.; Flegel, T.; et al. Muscular dystrophy in female dogs. J. Vet. Intern. Med. 2001, 15, 240–244. [Google Scholar] [CrossRef]
- Sarkozy, A.; Foley, A.R.; Zambon, A.A.; Bönnemann, C.G.; Muntoni, F. LAMA2-related dystrophies: Clinical phenotypes, disease biomarkers, and clinical trial readiness. Front. Mol. Neurosci. 2020, 13, 123. [Google Scholar] [CrossRef] [PubMed]
- Miner, J.H. Laminins and their roles in mammals. Microsc. Res. Tech. 2008, 71, 349–356. [Google Scholar] [CrossRef]
- Ehrig, K.; Leivo, I.; Argraves, W.S.; Ruoslahti, E.; Engvall, E. Merosin, a tissue-specific basement membrane protein, is a laminin-like protein. Proc. Natl. Acad. Sci. USA 1990, 87, 3264–3268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jimenez-Mallebrera, C.; Brown, S.C.; Sewry, C.A.; Muntoni, F. Congenital muscular dystrophy: Molecular and cellular aspects. Cell. Mol. Life Sci. 2005, 62, 809–823. [Google Scholar] [CrossRef]
- Holmberg, J.; Durbeej, M. Laminin-211 in skeletal muscle function. Cell Adhes. Migr. 2013, 7, 111–121. [Google Scholar] [CrossRef] [Green Version]
- Helbling-Leclerc, A.; Zhang, X.; Topaloglu, H.; Cruaud, C.; Tesson, F.; Weissenbach, J.; Tomé, F.M.S.; Schwartz, K.; Fardeau, M.; Tryggvason, K.; et al. Mutations in the laminin α2–chain gene (LAMA2) cause merosin–deficient congenital muscular dystrophy. Nat. Genet. 1995, 11, 216–218. [Google Scholar] [CrossRef]
- Jones, K.J.; Morgan, G.; Johnston, H.; Tobias, V.; Ouvrier, R.A.; Wilkinson, I.; North, K.N. The expanding phenotype of laminin alpha2 chain (merosin) abnormalities: Case series and review. J. Med. Genet. 2001, 38, 649–657. [Google Scholar] [CrossRef] [PubMed]
- Allamand, V.; Guicheney, P. Merosin-deficient congenital muscular dystrophy, autosomal recessive (MDC1A, MIM#156225, LAMA2 gene coding for α2 chain of laminin). Eur. J. Hum. Genet. 2002, 10, 91–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geranmayeh, F.; Clement, E.; Feng, L.H.; Sewry, C.; Pagan, J.; Mein, R.; Abbs, S.; Brueton, L.; Childs, A.-M.; Jungbluth, H.; et al. Genotype–phenotype correlation in a large population of muscular dystrophy patients with LAMA2 mutations. Neuromuscul. Disord. 2010, 20, 241–250. [Google Scholar] [CrossRef] [PubMed]
- Bönnemann, C.G.; Wang, C.H.; Quijano-Roy, S.; Deconinck, N.; Bertini, E.; Ferreiro, A.; Muntoni, F.; Sewry, C.; Béroud, C.; Mathews, K.D.; et al. Diagnostic approach to the congenital muscular dystrophies. Neuromuscul. Disord. 2014, 24, 289–311. [Google Scholar] [CrossRef] [PubMed]
- Xiong, H.; Tan, D.; Wang, S.; Song, S.; Yang, H.; Gao, K.; Liu, A.; Jiao, H.; Mao, B.; Ding, J.; et al. Genotype/phenotype analysis in Chinese laminin-α2 deficient congenital muscular dystrophy patients. Clin. Genet. 2015, 87, 233–243. [Google Scholar] [CrossRef]
- Oliveira, J.; Gruber, A.; Cardoso, M.; Taipa, R.; Fineza, I.; Gonçalves, A.; Laner, A.; Winder, T.L.; Schroeder, J.; Rath, J.; et al. LAMA2 gene mutation update: Toward a more comprehensive picture of the laminin-α2 variome and its related phenotypes. Hum. Mutat. 2018, 39, 1314–1337. [Google Scholar] [CrossRef]
- Yurchenco, P.D.; McKee, K.K.; Reinhard, J.R.; Rüegg, M.A. Laminin-deficient muscular dystrophy: Molecular pathogenesis and structural repair strategies. Matrix Biol. 2018, 71–72, 174–187. [Google Scholar] [CrossRef]
- Previtali, S.C.; Zambon, A.A. LAMA2 neuropathies: Human findings and pathomechanisms from mouse models. Front. Mol. Neurosci. 2020, 13, 60. [Google Scholar] [CrossRef]
- O’Brien, D.P.; Johnson, G.C.; Liu, L.A.; Guo, L.T.; Engvall, E.; Powell, H.C.; Shelton, G.D. Laminin alpha 2 (merosin)-deficient muscular dystrophy and demyelinating neuropathy in two cats. J. Neurol. Sci. 2001, 189, 37–43. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Gawlik, K.I.; Durbeej, M. A family of laminin α2 chain-deficient mouse mutants: Advancing the research on LAMA2-CMD. Front. Mol. Neurosci. 2020, 13, 59. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Filtering Step | Homozygous Variants | Heterozygous Variants |
|---|---|---|
| All variants in the affected dog | 2,819,563 | 3,360,003 |
| Private variants | 1476 | 8957 |
| Protein-changing private variants | 6 | 68 |
| in 39 known CMD candidate genes [1] | 1 | 0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Christen, M.; Indzhova, V.; Guo, L.T.; Jagannathan, V.; Leeb, T.; Shelton, G.D.; Brocal, J. LAMA2 Nonsense Variant in an Italian Greyhound with Congenital Muscular Dystrophy. Genes 2021, 12, 1823. https://doi.org/10.3390/genes12111823
Christen M, Indzhova V, Guo LT, Jagannathan V, Leeb T, Shelton GD, Brocal J. LAMA2 Nonsense Variant in an Italian Greyhound with Congenital Muscular Dystrophy. Genes. 2021; 12(11):1823. https://doi.org/10.3390/genes12111823
Chicago/Turabian StyleChristen, Matthias, Victoria Indzhova, Ling T. Guo, Vidhya Jagannathan, Tosso Leeb, G. Diane Shelton, and Josep Brocal. 2021. "LAMA2 Nonsense Variant in an Italian Greyhound with Congenital Muscular Dystrophy" Genes 12, no. 11: 1823. https://doi.org/10.3390/genes12111823