
Clinical and Genetic Study of X-Linked Juvenile Retinoschisis in the Czech Population

Abstract
:1. Introduction
2. Materials and Methods
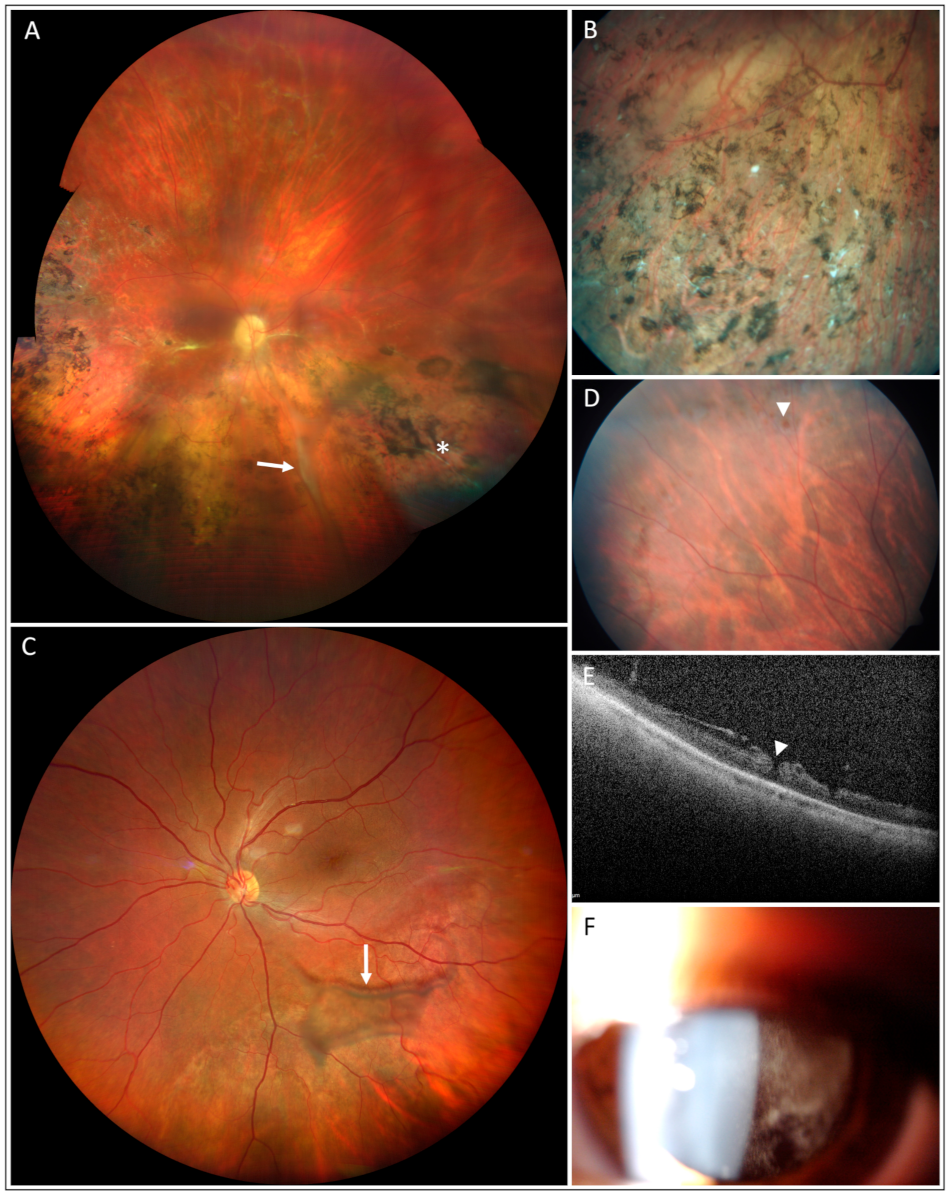
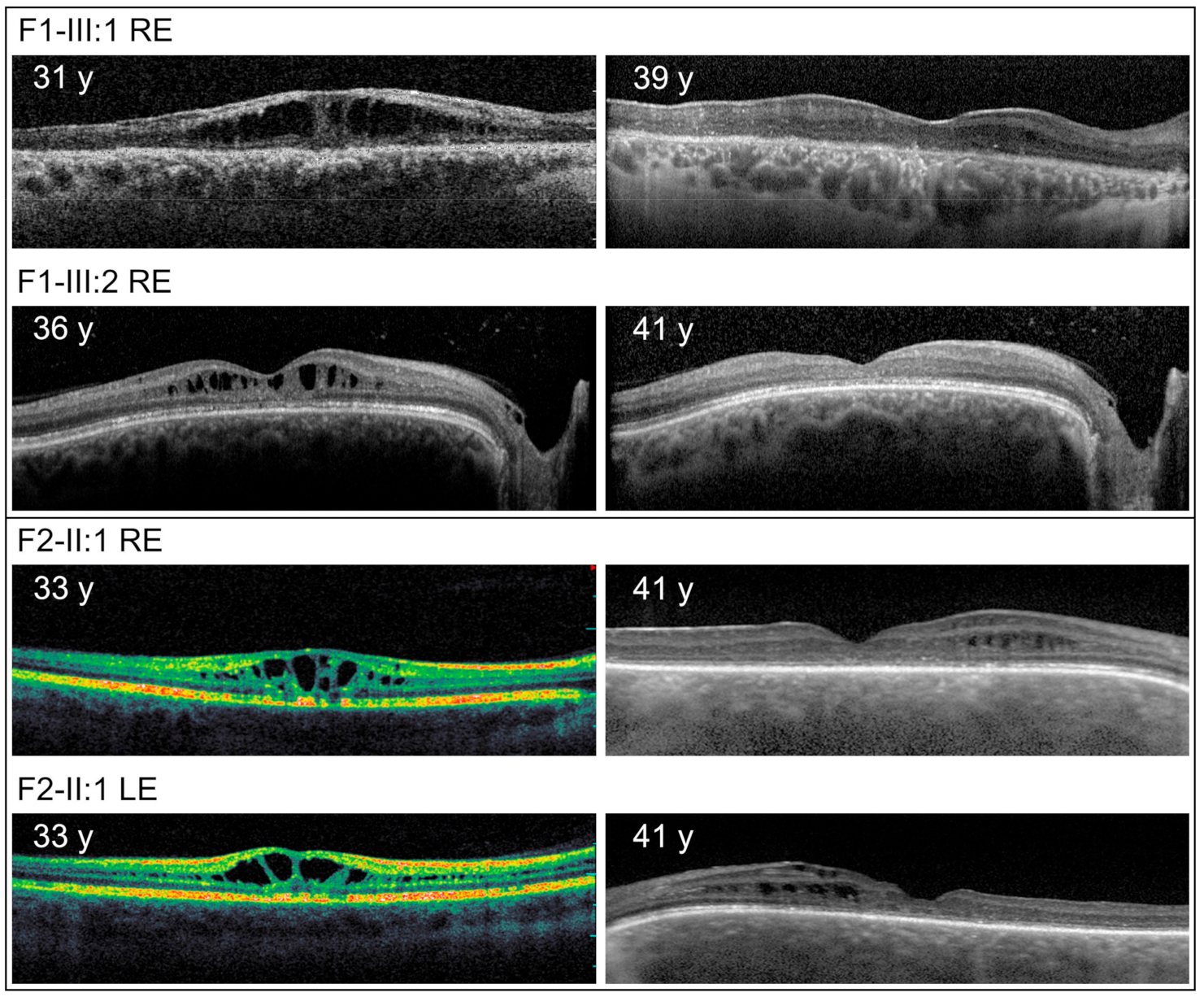
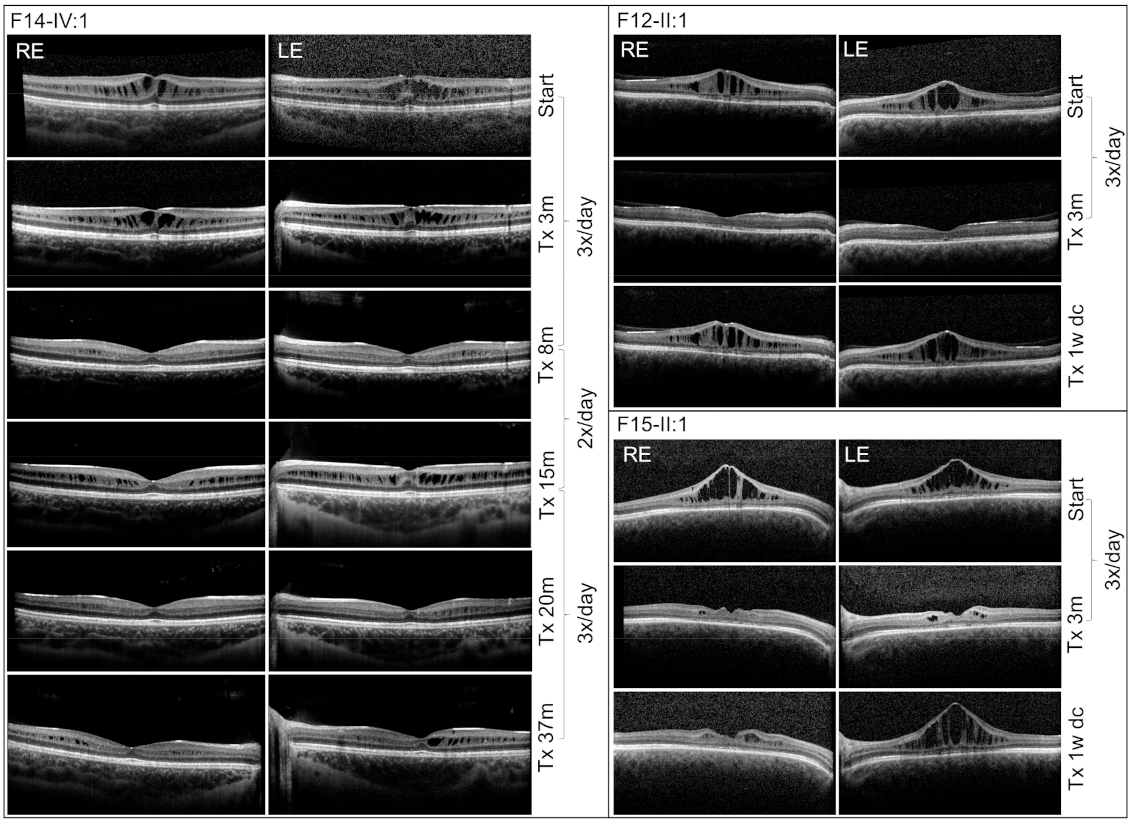
3. Results
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Molday, R.S.; Kellner, U.; Weber, B.H. X-linked juvenile retinoschisis: Clinical diagnosis, genetic analysis, and molecular mechanisms. Prog. Retin. Eye Res. 2012, 31, 195–212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- George, N.D.; Yates, J.R.; Moore, A.T. X linked retinoschisis. Br. J. Ophthalmol. 1995, 79, 697–702. [Google Scholar] [CrossRef] [Green Version]
- Hu, Q.R.; Huang, L.Z.; Chen, X.L.; Xia, H.K.; Li, T.Q.; Li, X.X. Genetic analysis and clinical features of X-linked retinoschisis in Chinese patients. Sci. Rep. 2017, 7, 44060. [Google Scholar] [CrossRef]
- Hinds, A.M.; Fahim, A.; Moore, A.T.; Wong, S.C.; Michaelides, M. Bullous X linked retinoschisis: Clinical features and prognosis. Br. J. Ophthalmol. 2018, 102, 622–624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- George, N.D.; Yates, J.R.; Moore, A.T. Clinical features in affected males with X-linked retinoschisis. Arch. Ophthalmol. 1996, 114, 274–280. [Google Scholar] [CrossRef] [PubMed]
- Kellner, U.; Brümmer, S.; Foerster, M.H.; Wessing, A. X-linked congenital retinoschisis. Graefe’s Arch. Clin. Exp. Ophthalmol. 1990, 228, 432–437. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.K.; Liu, L.; Chen, H.M.; Tsai, S.; Chang, T.C.; Tsai, T.H.; Yang, C.M.; Chao, A.N.; Chen, K.J.; Kao, L.Y.; et al. Clinical presentations of X-linked retinoschisis in Taiwanese patients confirmed with genetic sequencing. Mol. Vis. 2015, 21, 487–501. [Google Scholar] [PubMed]
- Hotta, Y.; Nakamura, M.; Okamoto, Y.; Nomura, R.; Terasaki, H.; Miyake, Y. Different mutation of the XLRS1 gene causes juvenile retinoschisis with retinal white flecks. Br. J. Ophthalmol. 2001, 85, 238–239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fahim, A.T.; Ali, N.; Blachley, T.; Michaelides, M. Peripheral fundus findings in X-linked retinoschisis. Br. J. Ophthalmol. 2017, 101, 1555–1559. [Google Scholar] [CrossRef] [PubMed]
- Apushkin, M.A.; Fishman, G.A.; Rajagopalan, A.S. Fundus findings and longitudinal study of visual acuity loss in patients with X-linked retinoschisis. Retina 2005, 25, 612–618. [Google Scholar] [CrossRef]
- Pimenides, D.; George, N.D.; Yates, J.R.; Bradshaw, K.; Roberts, S.A.; Moore, A.T.; Trump, D. X-linked retinoschisis: Clinical phenotype and RS1 genotype in 86 UK patients. J. Med. Genet. 2005, 42, e35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khandhadia, S.; Trump, D.; Menon, G.; Lotery, A.J. X-linked retinoschisis maculopathy treated with topical dorzolamide, and relationship to genotype. Eye 2011, 25, 922–928. [Google Scholar] [CrossRef] [Green Version]
- Collison, F.T.; Genead, M.A.; Fishman, G.A.; Stone, E.M. Resolution of mid-peripheral schisis in x-linked retinoschisis with the use of dorzolamide. Ophthalmic Genet. 2014, 35, 125–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thobani, A.; Fishman, G.A. The use of carbonic anhydrase inhibitors in the retreatment of cystic macular lesions in retinitis pigmentosa and X-linked retinoschisis. Retina 2011, 31, 312–315. [Google Scholar] [CrossRef]
- Sauer, C.G.; Gehrig, A.; Warneke-Wittstock, R.; Marquardt, A.; Ewing, C.C.; Gibson, A.; Lorenz, B.; Jurklies, B.; Weber, B.H. Positional cloning of the gene associated with X-linked juvenile retinoschisis. Nat. Genet. 1997, 17, 164–170. [Google Scholar] [CrossRef] [PubMed]
- Gehrig, A.; Weber, B.H.; Lorenz, B.; Andrassi, M. First molecular evidence for a de novo mutation in RS1 (XLRS1) associated with X linked juvenile retinoschisis. J. Med. Genet. 1999, 36, 932–934. [Google Scholar] [PubMed]
- Khan, N.W.; Jamison, J.A.; Kemp, J.A.; Sieving, P.A. Analysis of photoreceptor function and inner retinal activity in juvenile X-linked retinoschisis. Vision Res. 2001, 41, 3931–3942. [Google Scholar] [CrossRef] [Green Version]
- Molday, L.L.; Hicks, D.; Sauer, C.G.; Weber, B.H.; Molday, R.S. Expression of X-linked retinoschisis protein RS1 in photoreceptor and bipolar cells. Investig. Ophthalmol. Vis. Sci. 2001, 42, 816–825. [Google Scholar]
- Xiao, S.; Sun, W.; Xiao, X.; Li, S.; Luo, H.; Jia, X.; Ouyang, J.; Li, X.; Wang, Y.; Jiang, Y.; et al. Clinical and genetic features of retinoschisis in 120 families with RS1 mutations. Br. J. Ophthalmol. 2021. [Google Scholar] [CrossRef]
- Kim, D.Y.; Mukai, S. X-linked juvenile retinoschisis (XLRS): A review of genotype-phenotype relationships. Semin. Ophthalmol. 2013, 28, 392–396. [Google Scholar] [CrossRef]
- McCulloch, D.L.; Marmor, M.F.; Brigell, M.G.; Hamilton, R.; Holder, G.E.; Tzekov, R.; Bach, M. ISCEV Standard for full-field clinical electroretinography (2015 update). Doc. Ophthalmol. 2015, 130, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Yi, J.; Li, S.; Jia, X.; Xiao, X.; Wang, P.; Guo, X.; Zhang, Q. Novel RS1 mutations associated with X-linked juvenile retinoschisis. Int. J. Mol. Med. 2012, 29, 644–648. [Google Scholar] [CrossRef] [Green Version]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- The Retinoschisis Consortium. Functional implications of the spectrum of mutations found in 234 cases with X-linked juvenile retinoschisis. Hum. Mol. Genet. 1998, 7, 1185–1192. [Google Scholar] [CrossRef] [Green Version]
- Huang, L.; Sun, L.; Wang, Z.; Chen, C.; Wang, P.; Sun, W.; Luo, X.; Ding, X. Clinical manifestation and genetic analysis in Chinese early onset X-linked retinoschisis. Mol. Genet. Genomic Med. 2020, 8, e1421. [Google Scholar] [CrossRef] [PubMed]
- Hotta, Y.; Fujiki, K.; Hayakawa, M.; Ohta, T.; Fujimaki, T.; Tamaki, K.; Yokoyama, T.; Kanai, A.; Hirakata, A.; Hida, T.; et al. Japanese juvenile retinoschisis is caused by mutations of the XLRS1 gene. Hum. Genet. 1998, 103, 142–144. [Google Scholar] [CrossRef] [PubMed]
- Hiriyanna, K.T.; Bingham, E.L.; Yashar, B.M.; Ayyagari, R.; Fishman, G.; Small, K.W.; Weinberg, D.V.; Weleber, R.G.; Lewis, R.A.; Andreasson, S.; et al. Novel mutations in XLRS1 causing retinoschisis, including first evidence of putative leader sequence change. Hum. Mutat. 1999, 14, 423–427. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.Y.; Ko, H.S.; Yu, Y.S.; Hwang, J.M.; Lee, J.J.; Kim, S.Y.; Kim, J.Y.; Seong, M.W.; Park, K.H.; Park, S.S. Molecular genetic characteristics of X-linked retinoschisis in Koreans. Mol. Vis. 2009, 15, 833–843. [Google Scholar]
- Riveiro-Alvarez, R.; Trujillo-Tiebas, M.J.; Gimenez-Pardo, A.; Garcia-Hoyos, M.; Lopez-Martinez, M.A.; Aguirre-Lamban, J.; Garcia-Sandoval, B.; Vazquez-Fernandez del Pozo, S.; Cantalapiedra, D.; Avila-Fernandez, A.; et al. Correlation of genetic and clinical findings in Spanish patients with X-linked juvenile retinoschisis. Investig. Ophthalmol. Vis. Sci. 2009, 50, 4342–4350. [Google Scholar] [CrossRef]
- Mashima, Y.; Shinoda, K.; Ishida, S.; Ozawa, Y.; Kudoh, J.; Iwata, T.; Oguchi, Y.; Shimizu, N. Identification of four novel mutations of the XLRS1 gene in Japanese patients with X-linked juvenile retinoschisis. Mutation in brief no. 234. Online. Hum. Mutat. 1999, 13, 338. [Google Scholar] [CrossRef]
- Inoue, Y.; Yamamoto, S.; Okada, M.; Tsujikawa, M.; Inoue, T.; Okada, A.A.; Kusaka, S.; Saito, Y.; Wakabayashi, K.; Miyake, Y.; et al. X-linked retinoschisis with point mutations in the XLRS1 gene. Arch. Ophthalmol. 2000, 118, 93–96. [Google Scholar] [CrossRef] [Green Version]
- Curat, C.A.; Eck, M.; Dervillez, X.; Vogel, W.F. Mapping of epitopes in discoidin domain receptor 1 critical for collagen binding. J. Biol. Chem. 2001, 276, 45952–45958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, T.; Waters, C.T.; Rothman, A.M.; Jakins, T.J.; Römisch, K.; Trump, D. Intracellular retention of mutant retinoschisin is the pathological mechanism underlying X-linked retinoschisis. Hum. Mol. Genet. 2002, 11, 3097–3105. [Google Scholar] [CrossRef] [PubMed]
- Robson, A.G.; Mengher, L.S.; Tan, M.H.; Moore, A.T. An unusual fundus phenotype of inner retinal sheen in X-linked retinoschisis. Eye 2009, 23, 1876–1878. [Google Scholar] [CrossRef]
- Vijayasarathy, C.; Ziccardi, L.; Zeng, Y.; Smaoui, N.; Caruso, R.C.; Sieving, P.A. Null retinoschisin-protein expression from an RS1 c354del1-ins18 mutation causing progressive and severe XLRS in a cross-sectional family study. Investig. Ophthalmol. Vis. Sci. 2009, 50, 5375–5383. [Google Scholar] [CrossRef]
- Kjellström, S.; Vijayasarathy, C.; Ponjavic, V.; Sieving, P.A.; Andréasson, S. Long-term 12 year follow-up of X-linked congenital retinoschisis. Ophthalmic. Genet. 2010, 31, 114–125. [Google Scholar] [CrossRef]
- Bennett, L.D.; Wang, Y.Z.; Klein, M.; Pennesi, M.E.; Jayasundera, T.; Birch, D.G. Structure/Psychophysical Relationships in X-Linked Retinoschisis. Investig. Ophthalmol. Vis. Sci. 2016, 57, 332–337. [Google Scholar] [CrossRef] [Green Version]
- Stringa, F.; Tsamis, E.; Papayannis, A.; Chwiejczak, K.; Jalil, A.; Biswas, S.; Ahmad, H.; Stanga, P.E. Segmented swept source optical coherence tomography angiography assessment of the perifoveal vasculature in patients with X-linked juvenile retinoschisis: A serial case report. Int. Med. Case. Rep. J. 2017, 10, 329–335. [Google Scholar] [CrossRef] [Green Version]
- Padrón-Pérez, N.; Català-Mora, J.; Díaz, J.; Arias, L.; Prat, J.; Caminal, J.M. Swept-source and optical coherence tomography angiography in patients with X-linked retinoschisis. Eye 2018, 32, 707–715. [Google Scholar] [CrossRef]
- Mastropasqua, R.; Toto, L.; Di Antonio, L.; Parodi, M.B.; Sorino, L.; Antonucci, I.; Stuppia, L.; Di Nicola, M.; Mariotti, C. Optical Coherence Tomography Angiography Findings in X-Linked Retinoschisis. Ophthalmic Surg. Lasers Imaging Retina 2018, 49, e20–e31. [Google Scholar] [CrossRef] [PubMed]
- Han, I.C.; Whitmore, S.S.; Critser, D.B.; Lee, S.Y.; DeLuca, A.P.; Daggett, H.T.; Affatigato, L.M.; Mullins, R.F.; Tucker, B.A.; Drack, A.V.; et al. Wide-Field Swept-Source OCT and Angiography in X-Linked Retinoschisis. Ophthalmol. Retina 2019, 3, 178–185. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Family | DNA Level | Protein Level | ClinVar Interpretation/VCV Accession | References |
|---|---|---|---|---|
| F1, F2 | c.20del | p.(Gly7Alafs*119) | Not present | Novel |
| F3, F4, F15 | c.33_36del | p.(Leu11Phefs*114) | Pathogenic/VCV000098944.7 | [25] |
| F5 | c.187T>C | p.(Cys63Arg) | Not present | [26] |
| F6 | c.275G>A | p.(Trp92*) | Not present | Novel |
| F7, F8 | c.305G>A | p.(Arg102Gln) | Pathogenic/VCV000009896.11 | [11,25,27,28,29] |
| F9 | c.375_379del | p.(Asp126Glufs*16) | Not present | Novel |
| F10 | c.539C>A | p.(Ser180*) | Not present | Novel |
| F16 | c.421C>T | p.(Arg141Cys) | Pathogenic/VCV000098959.9 | [11,25,30] |
| F11, F17 | c.544C>T | p.(Arg182Cys) | Pathogenic/VCV000098986.3 | [25,29,31,32] |
| F12 | c.574C>T | p.(Pro192Ser) | Pathogenic/VCV000098990.4 | [15,25,32] |
| F13 | c.575_576insT | p.(Ile194Hisfs*70) | Not present | Novel # |
| F14 | c.637C>T | p.(Arg213Trp) | Pathogenic/VCV000099009.5 | [25,29,33,34] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kousal, B.; Hlavata, L.; Vlaskova, H.; Dvorakova, L.; Brichova, M.; Dubska, Z.; Langrova, H.; Vincent, A.L.; Dudakova, L.; Liskova, P. Clinical and Genetic Study of X-Linked Juvenile Retinoschisis in the Czech Population. Genes 2021, 12, 1816. https://doi.org/10.3390/genes12111816
Kousal B, Hlavata L, Vlaskova H, Dvorakova L, Brichova M, Dubska Z, Langrova H, Vincent AL, Dudakova L, Liskova P. Clinical and Genetic Study of X-Linked Juvenile Retinoschisis in the Czech Population. Genes. 2021; 12(11):1816. https://doi.org/10.3390/genes12111816
Chicago/Turabian StyleKousal, Bohdan, Lucia Hlavata, Hana Vlaskova, Lenka Dvorakova, Michaela Brichova, Zora Dubska, Hana Langrova, Andrea L. Vincent, Lubica Dudakova, and Petra Liskova. 2021. "Clinical and Genetic Study of X-Linked Juvenile Retinoschisis in the Czech Population" Genes 12, no. 11: 1816. https://doi.org/10.3390/genes12111816