
More Than a Moggy; A Population Genetics Analysis of the United Kingdom’s Non-Pedigree Cats

 , , ,
, , ,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Participants
2.2. Samples, DNA Extraction and Genotyping
2.3. Genotype Analysis
2.4. Population Ancestry and Structure Analysis
2.5. Linkage Disequilibrium Analysis
2.6. Homozygosity by Descent
3. Results
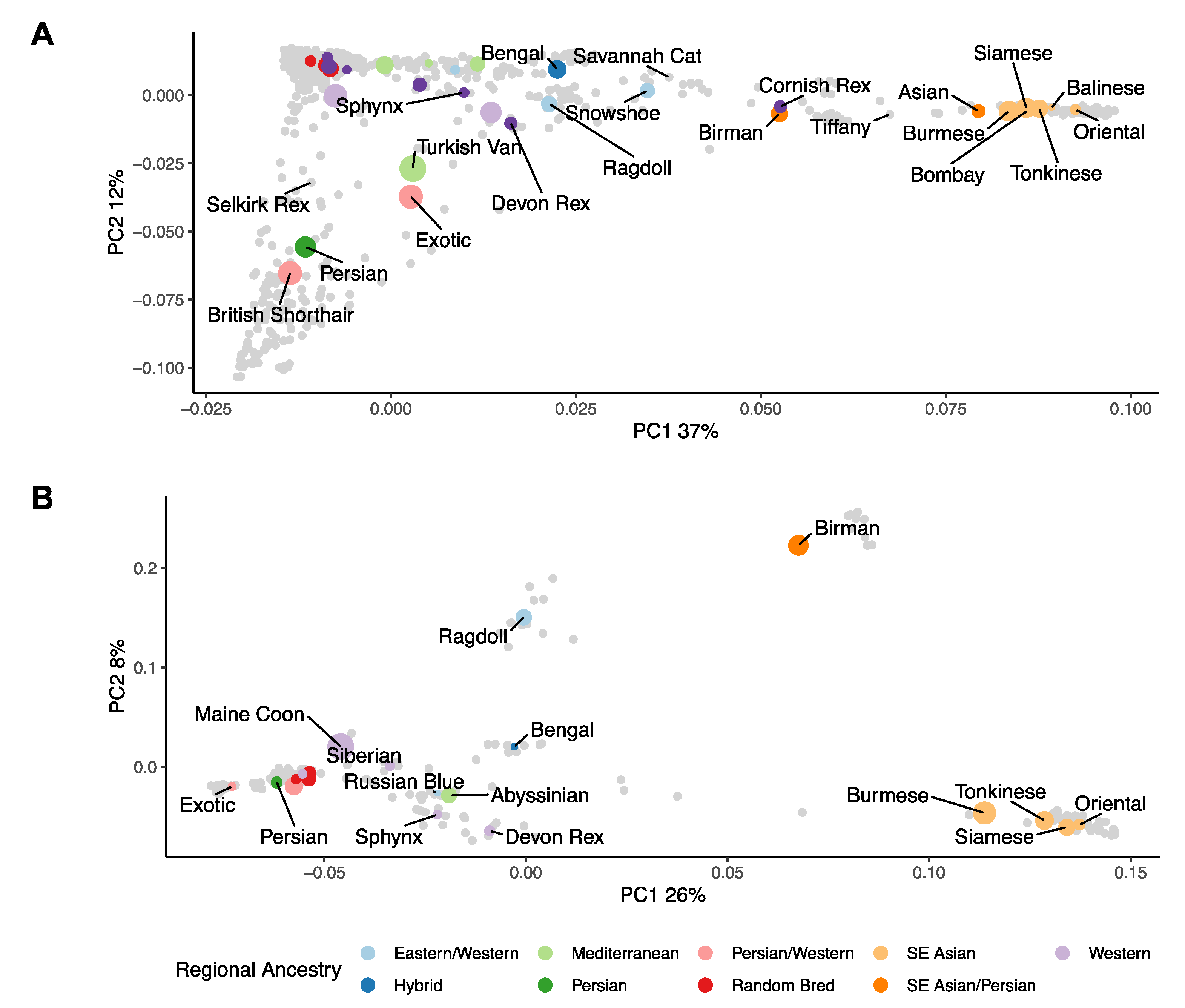
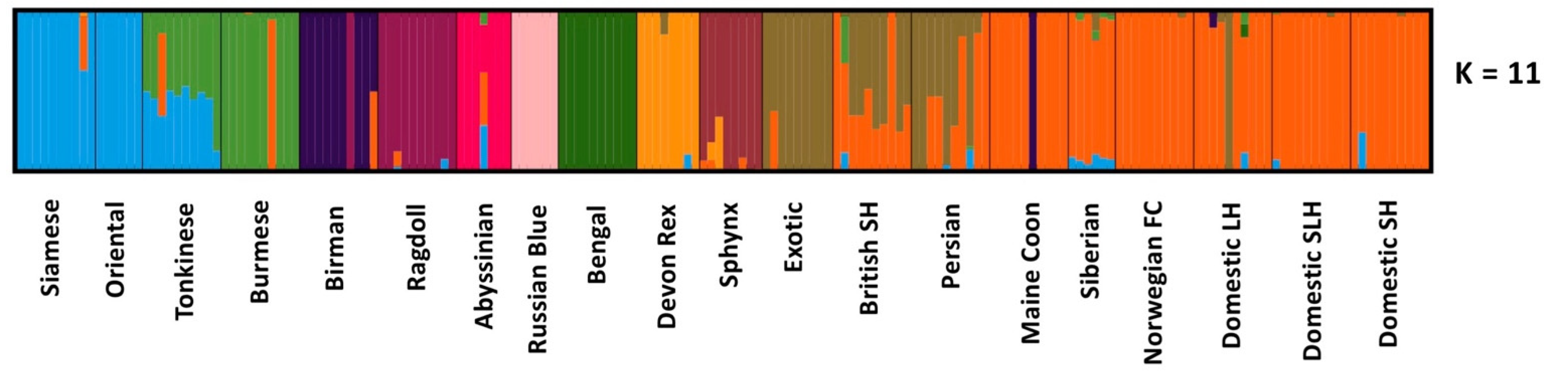
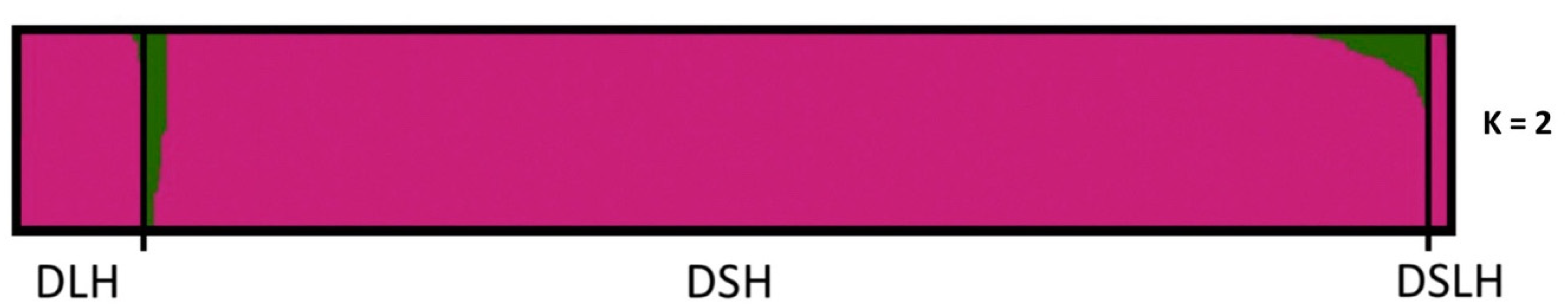
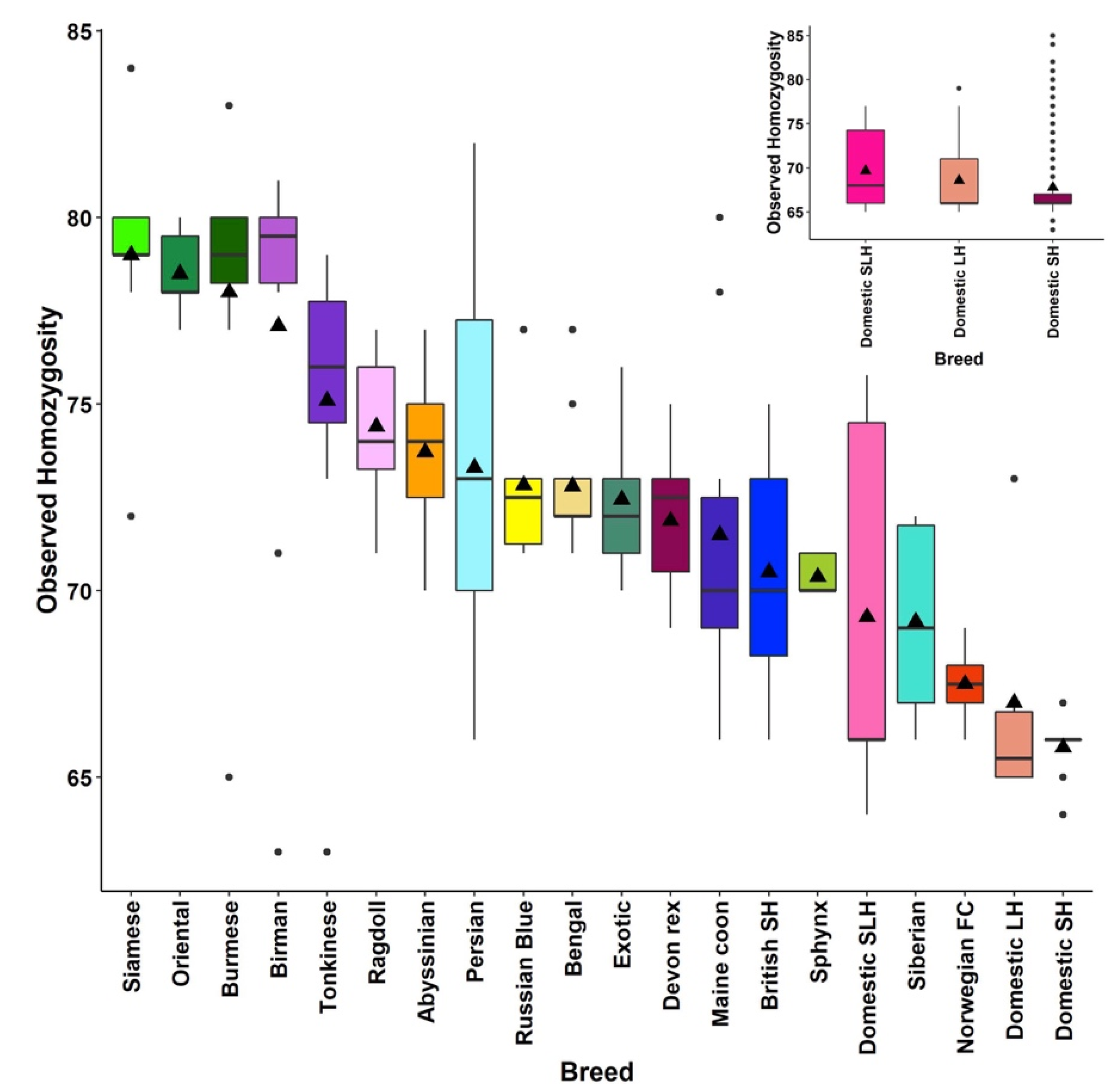
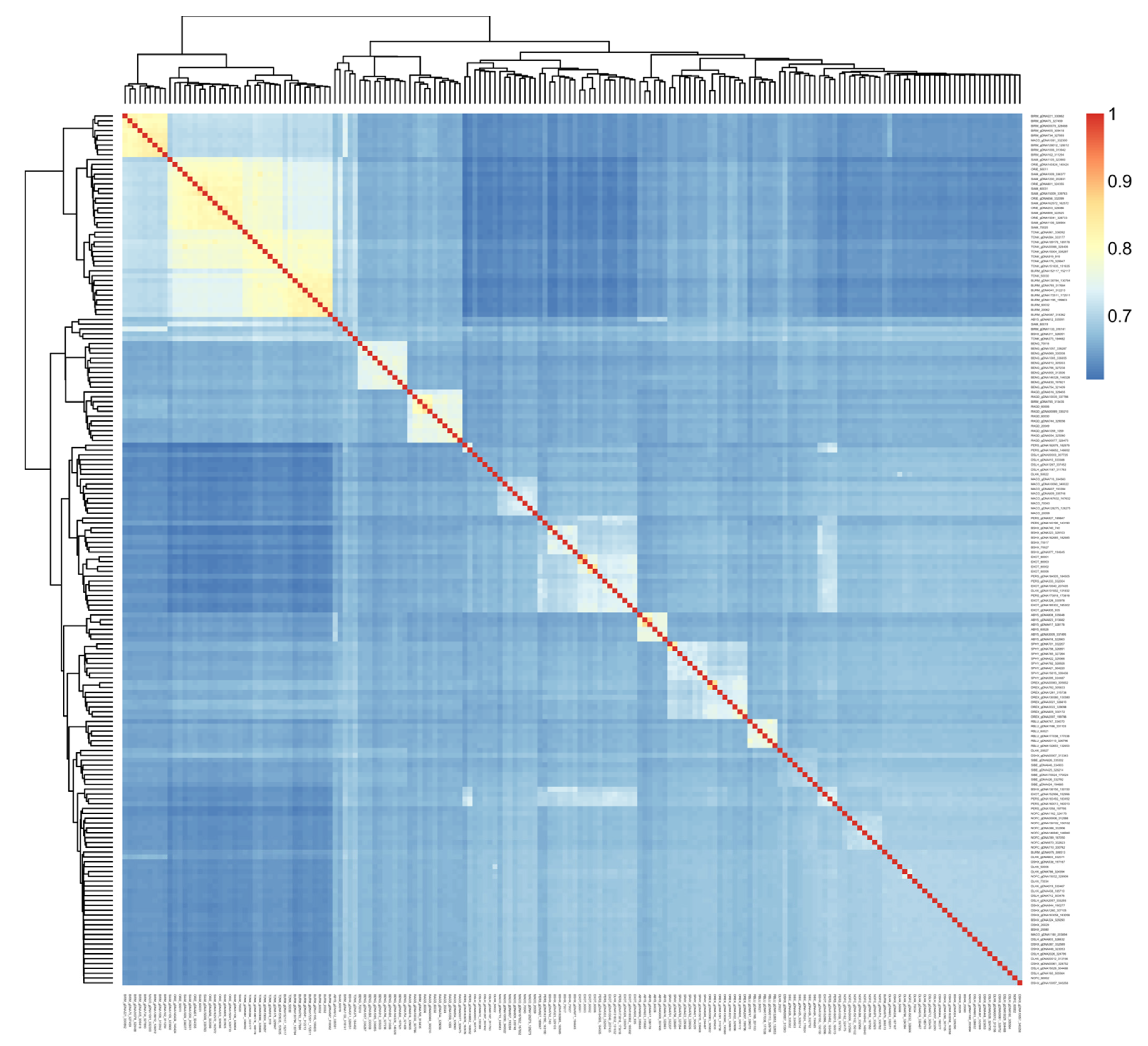
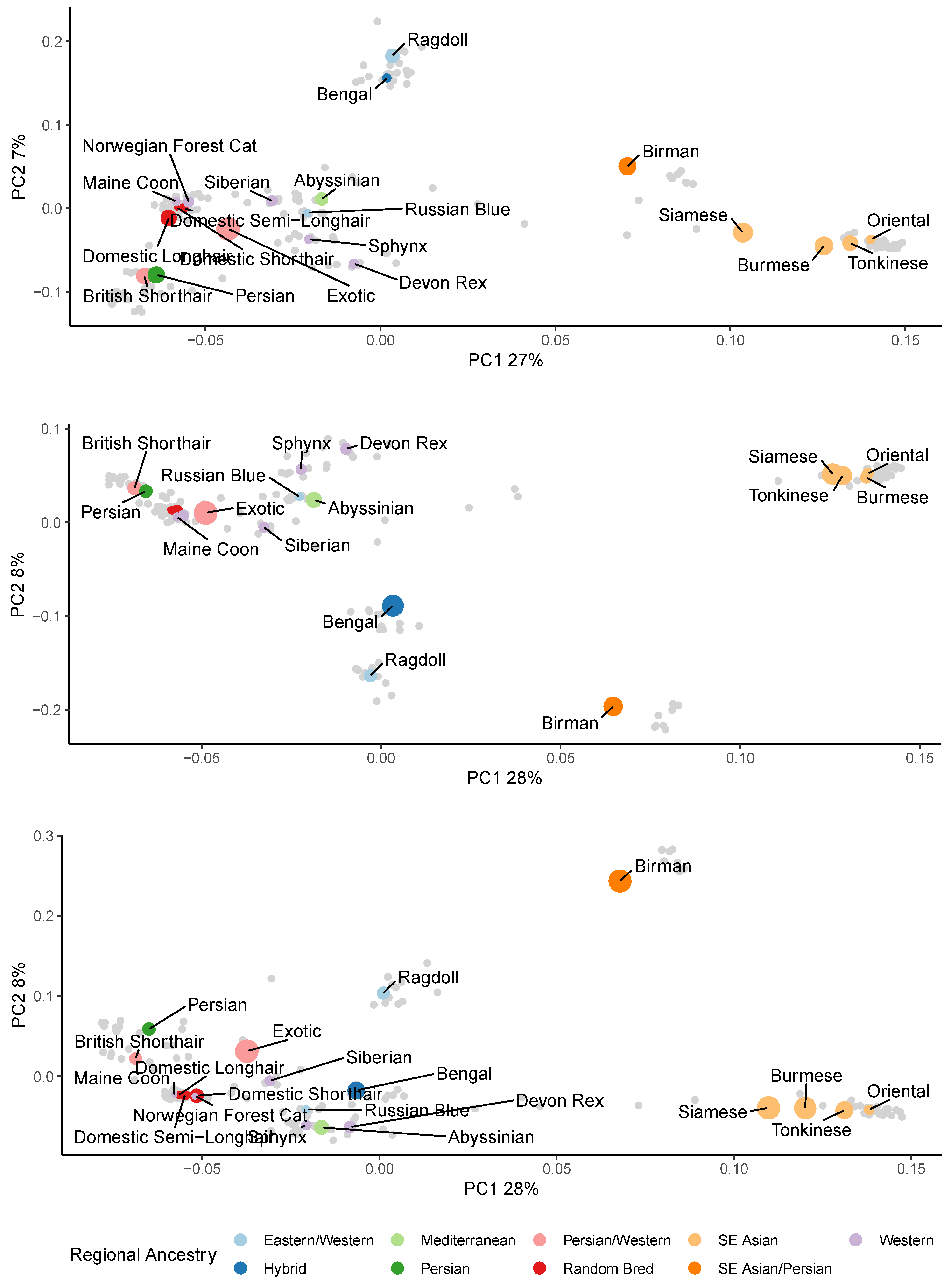
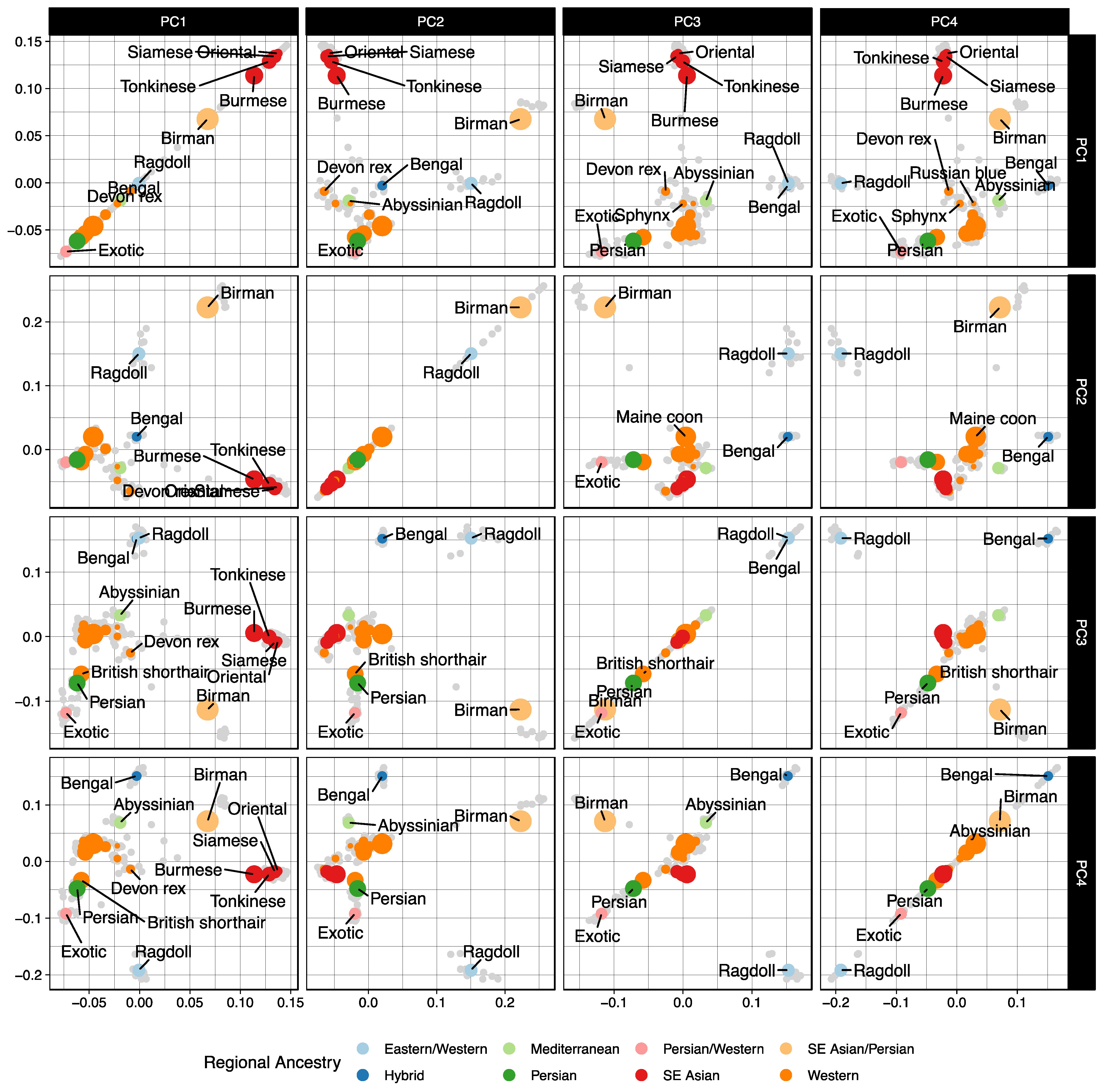
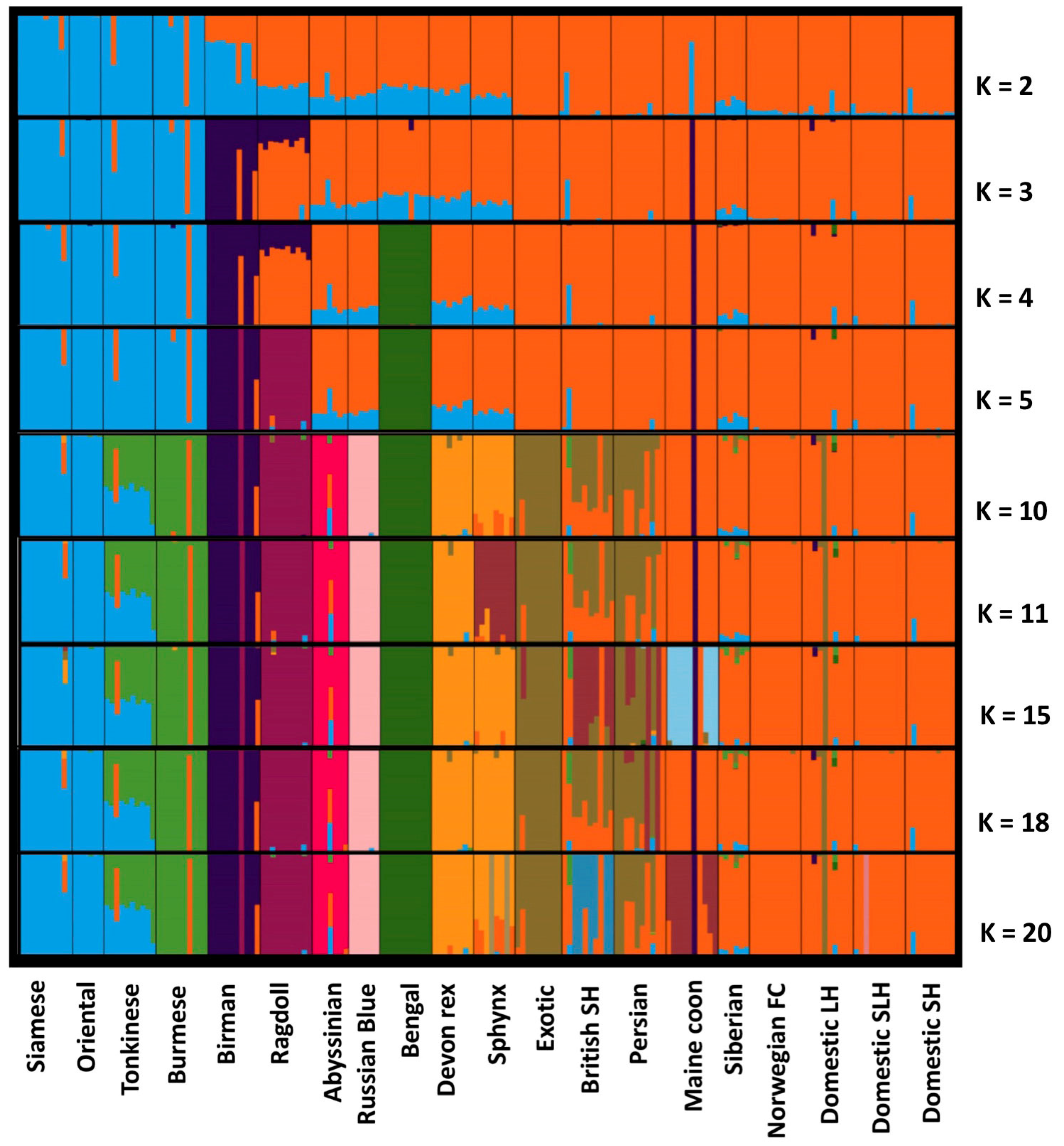
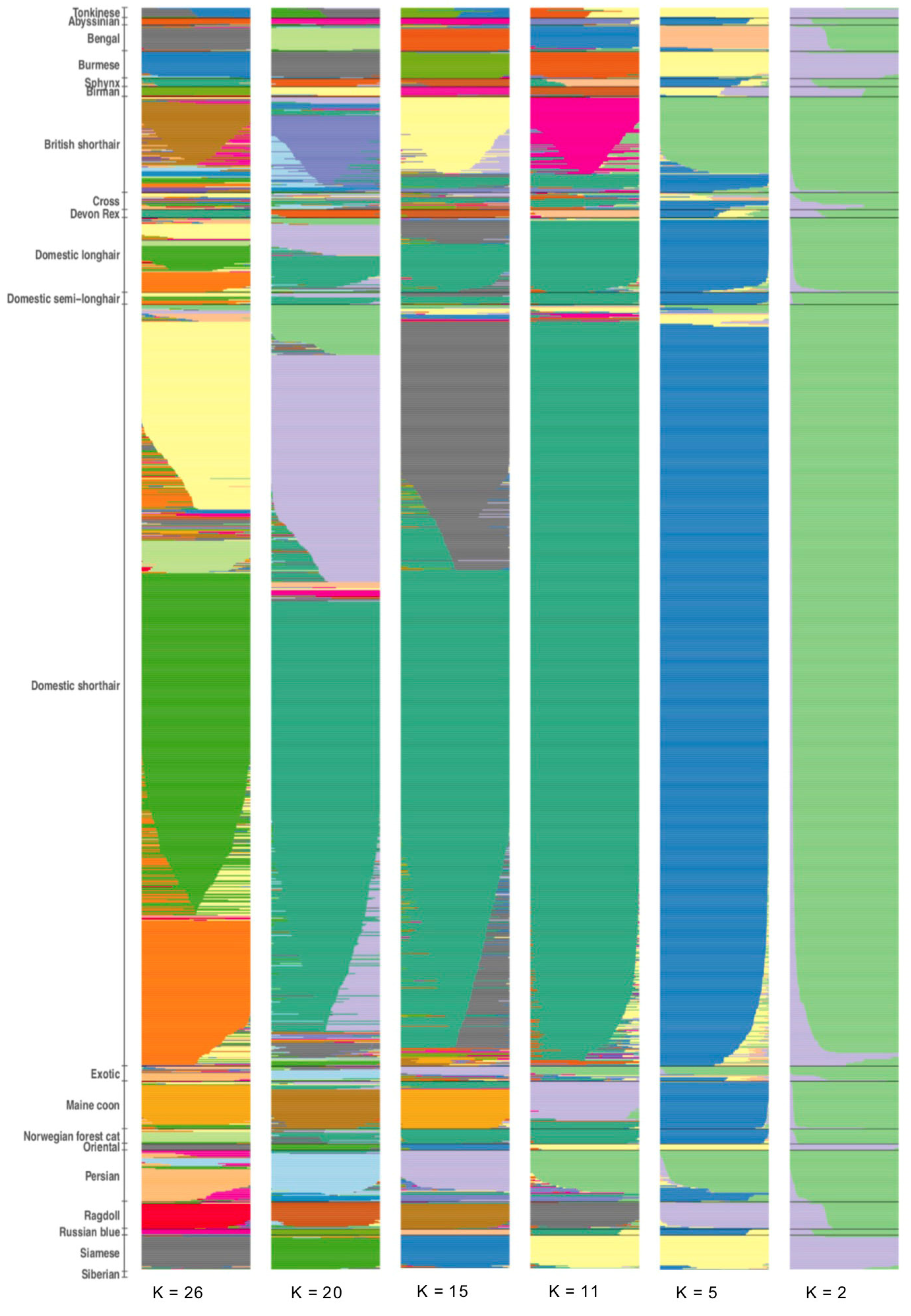
3.1. Population Ancestry and Structure
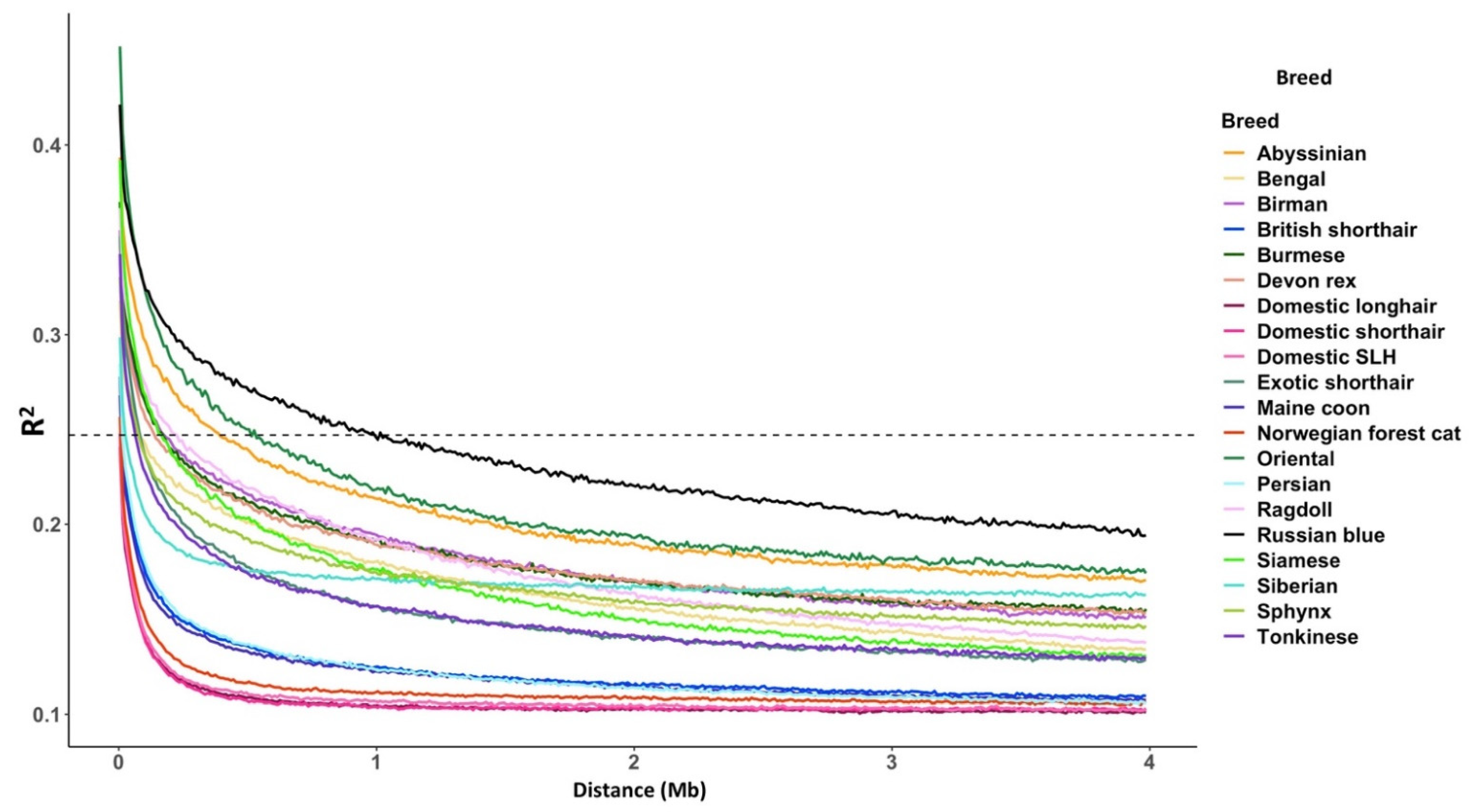
3.2. Linkage Disequilibrium
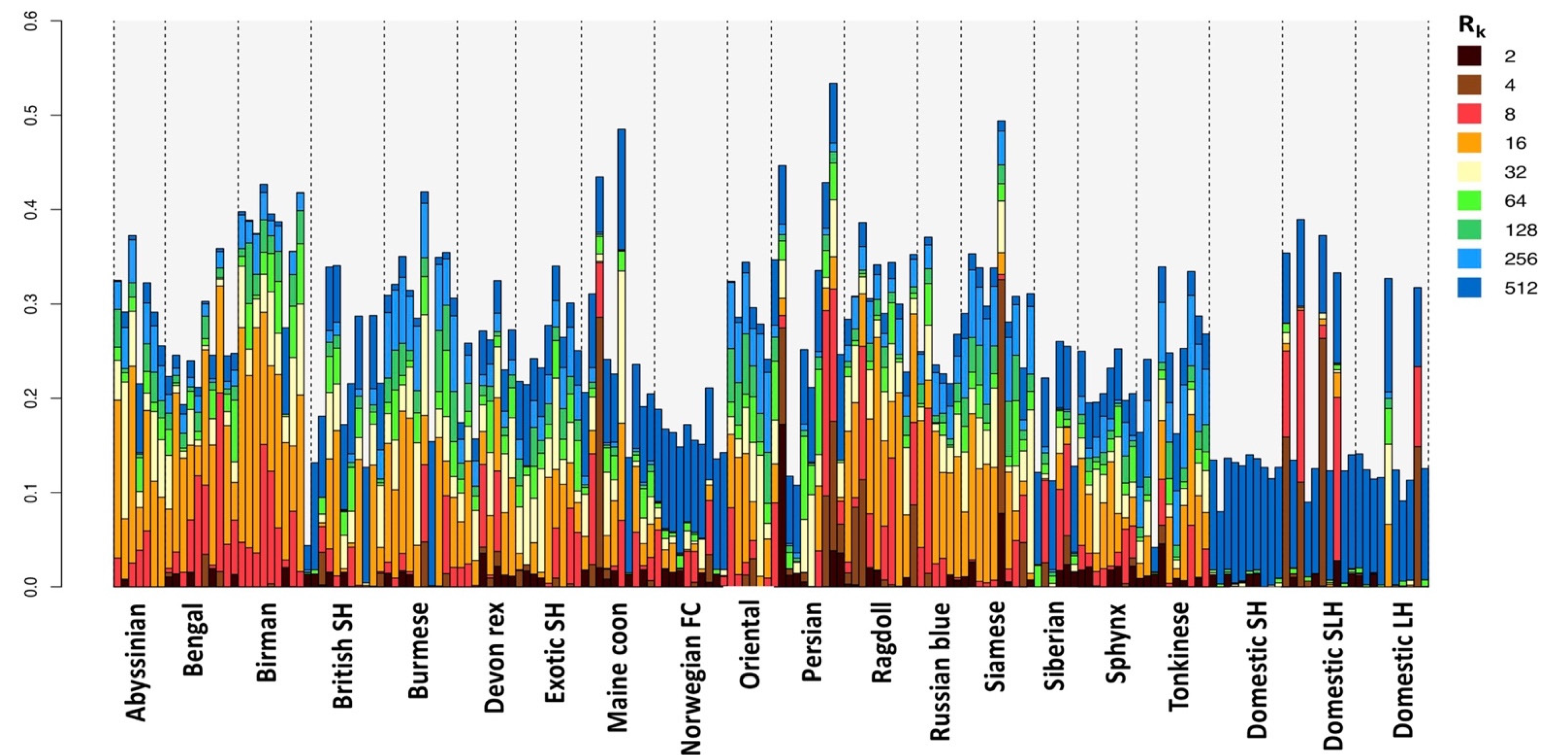
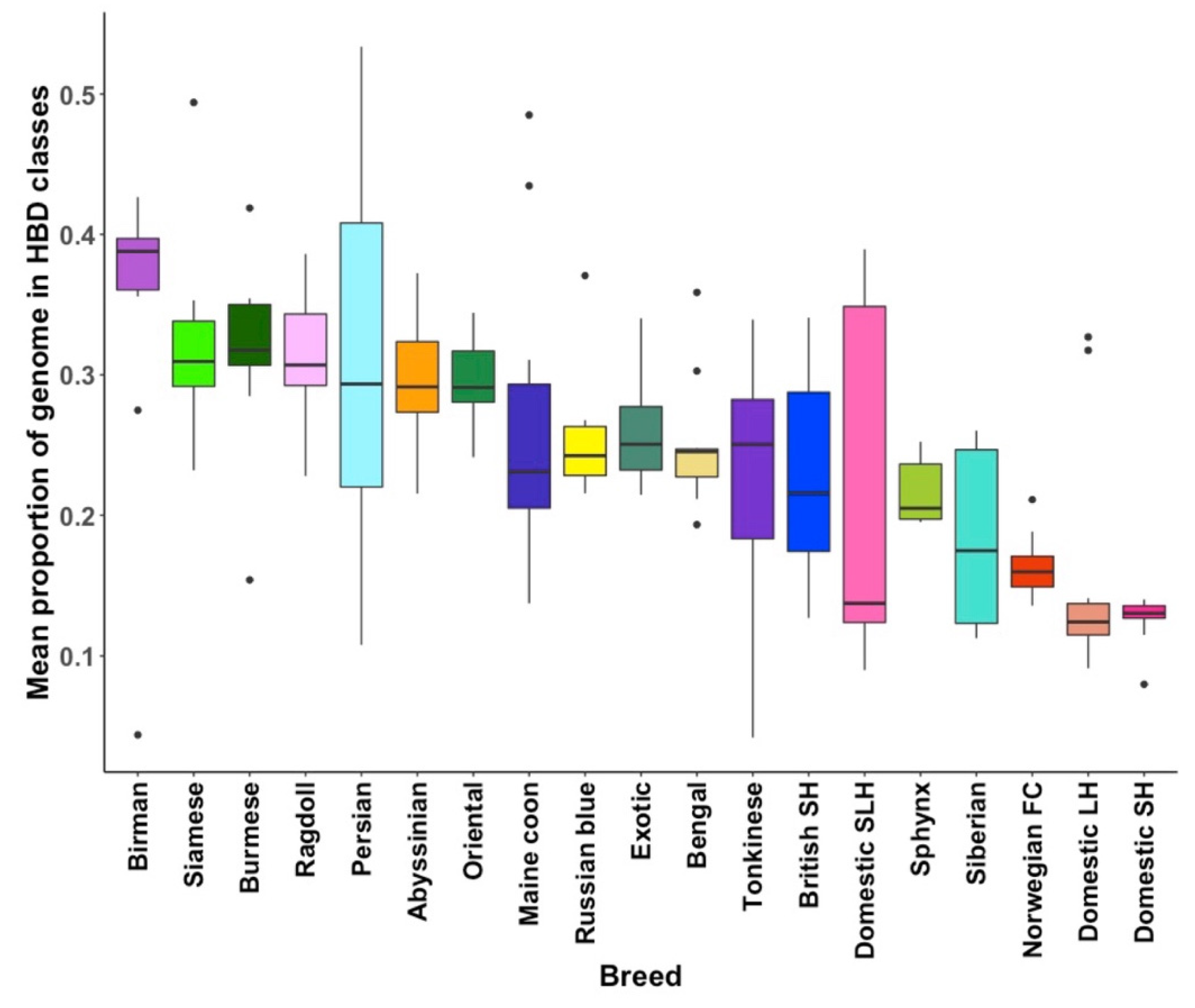
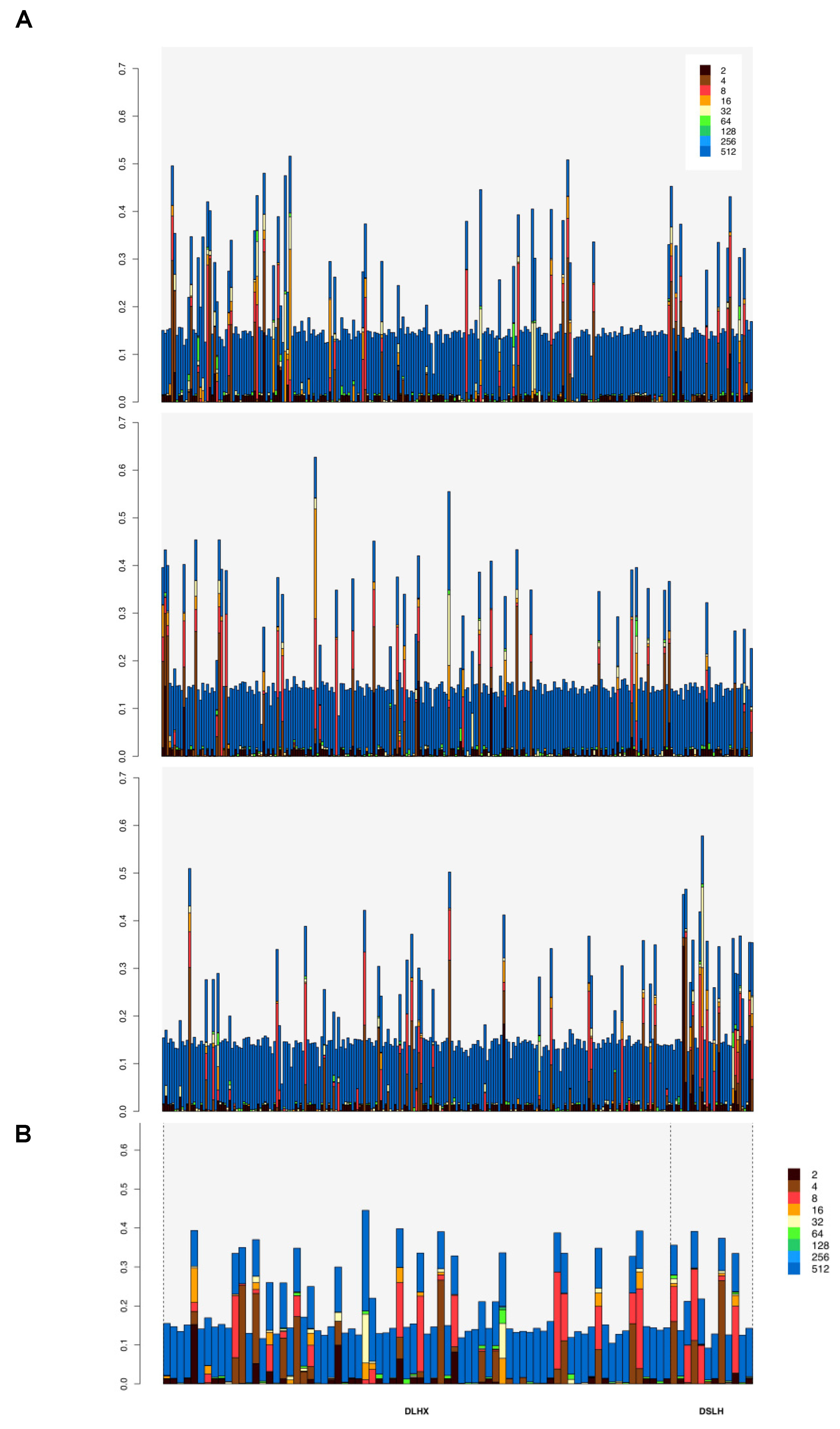
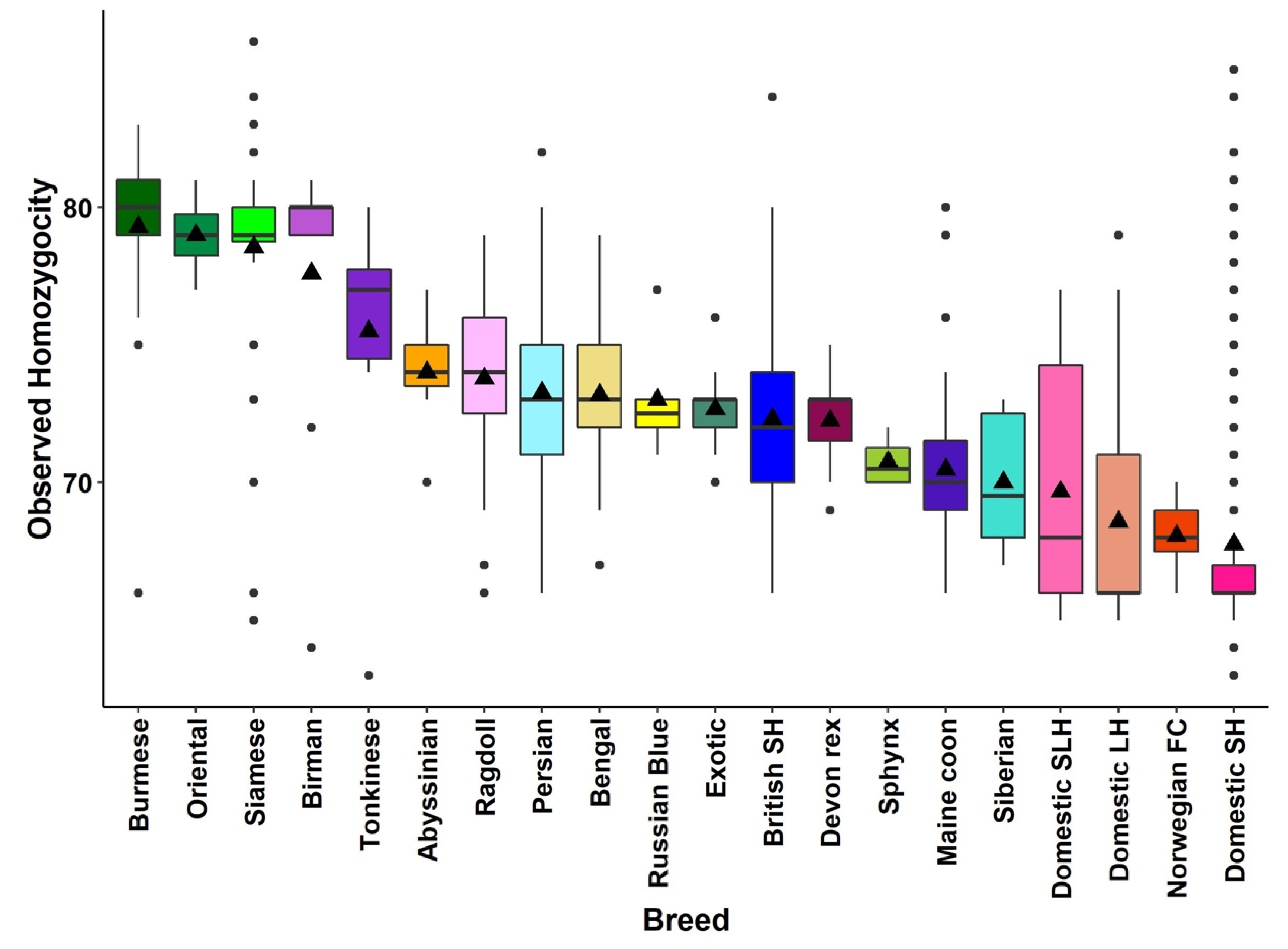
3.3. Homozygosity by Descent
4. Discussion
4.1. Breed Identification
4.2. The UK Population Compared to Previous Studies
4.3. What Are the UK’s Non-Pedigree Cats?
4.4. Health Implications for the UK’s Non-Pedigree Cats
4.5. SNV Array Performance
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A








References
- Statista. Cats in the United States Since 2000. Available online: https://www.statista.com/statistics/198102/cats-in-the-united-states-since-2000/#:~:text=According%20to%20a%20national%20pet,the%20United%20States%20in%202017 (accessed on 3 March 2021).
- PDSA. PDSA Animal Welfare (PAW) Report. Available online: https://www.pdsa.org.uk/media/10540/pdsa-paw-report-2020.pdf (accessed on 1 February 2021).
- O’Neill, D.; Church, D.; McGreevy, P.; Thomson, P.; Brodbelt, D. Prevalence of disorders recorded in cats attending primary-care veterinary practices in England. Veter. J. 2014, 202, 286–291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Neill, D.G.; Church, D.B.; McGreevy, P.; Thomson, P.; Brodbelt, D. Longevity and mortality of cats attending primary care veterinary practices in England. J. Feline Med. Surg. 2014, 17, 125–133. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, M.J.; Froenicke, L.; Baysac, K.C.; Billings, N.C.; Leutenegger, C.M.; Levy, A.M.; Longeri, M.; Niini, T.; Ozpinar, H.; Slater, M.R.; et al. The ascent of cat breeds: Genetic evaluations of breeds and worldwide random-bred populations. Genomics 2008, 91, 12–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurushima, J.D.; Lipinski, M.J.; Gandolfi, B.; Froenicke, L.; Grahn, J.C.; Grahn, R.A.; Lyons, L.A. Variation of cats under domestication: Genetic assignment of domestic cats to breeds and worldwide random-bred populations. Anim. Genet. 2012, 44, 311–324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vella, C.M.S.; Lorraine, M.; McGonagle, J.; Stanglein, T.W. Robinson’s Genetics for Cat Breeders and Veterinarians; Butterworth-Heinemann Ltd.: Oxford, UK, 1999. [Google Scholar]
- GCCF. GCCF Exotic Breeding Policy. Available online: https://www.gccfcats.org/Portals/0/Exotic.BP.pdf?ver=2017-03-29-132402-707 (accessed on 3 March 2021).
- Gandolfi, B.; Alhaddad, H.; Abdi, M.; Bach, L.H.; Creighton, E.K.; Davis, B.W.; Decker, J.E.; Dodman, N.H.; Ginns, E.I.; Grahn, J.C.; et al. Applications and efficiencies of the first cat 63K DNA array. Sci. Rep. 2018, 8. [Google Scholar] [CrossRef] [Green Version]
- Drögemüller, C.; Rüfenacht, S.; Wichert, B.; Leeb, T. Mutations within the FGF5 gene are associated with hair length in cats. Anim. Genet. 2007, 38, 218–221. [Google Scholar] [CrossRef]
- Kehler, J.S.; David, V.A.; Schäffer, A.A.; Bajema, K.; Eizirik, E.; Ryugo, D.K.; Hannah, S.S.; O’Brien, S.J.; Menotti-Raymond, M. Four independent mutations in the feline fibroblast growth factor 5 gene determine the long-haired phenotype in domestic cats. J. Hered. 2007, 98, 555–566. [Google Scholar] [CrossRef]
- Gandolfi, B.; Gruffydd-Jones, T.J.; Malik, R.; Cortes, A.; Jones, B.R.; Helps, C.; Prinzenberg, E.M.; Erhardt, G.; Lyons, L.A. First WNK4-Hypokalemia Animal Model Identified by Genome-Wide Association in Burmese Cats. PLoS ONE 2012, 7, e53173. [Google Scholar] [CrossRef] [Green Version]
- Gandolfi, B.; Grahn, R.A.; Creighton, E.K.; Williams, D.C.; Dickinson, P.J.; Sturges, B.K.; Guo, L.T.; Shelton, G.D.; Leegwater, P.A.J.; Longeri, M.; et al. COLQ variant associated with D evon R ex and S phynx feline hereditary myopathy. Anim. Genet. 2015, 46, 711–715. [Google Scholar] [CrossRef]
- Alhaddad, H.; Gandolfi, B.; Grahn, R.A.; Rah, H.-C.; Peterson, C.B.; Maggs, D.; Good, K.L.; Pedersen, N.C.; Lyons, L.A. Genome-wide association and linkage analyses localize a progressive retinal atrophy locus in Persian cats. Mamm. Genome 2014, 25, 354–362. [Google Scholar] [CrossRef]
- Samaha, G.; Wade, C.M.; Beatty, J.; Lyons, L.A.; Fleeman, L.M.; Haase, B. Mapping the genetic basis of diabetes mellitus in the Australian Burmese cat (Felis catus). Sci. Rep. 2020, 10. [Google Scholar] [CrossRef]
- Golovko, L.; Lyons, L.; Liu, H.; Sørensen, A.; Wehnert, S.; Pedersen, N.C. Genetic susceptibility to feline infectious peritonitis in Birman cats. Virus Res. 2013, 175, 58–63. [Google Scholar] [CrossRef]
- Bertolini, F.; Gandolfi, B.; Kim, E.S.; Haase, B.; Lyons, L.A.; Rothschild, M.F. Evidence of selection signatures that shape the Persian cat breed. Mamm. Genome 2016, 27, 144–155. [Google Scholar] [CrossRef]
- Marchant, T.W.; Johnson, E.J.; McTeir, L.; Johnson, C.I.; Gow, A.; Liuti, T.; Kuehn, D.; Svenson, K.; Bermingham, M.L.; Drögemüller, M.; et al. Canine Brachycephaly Is Associated with a Retrotransposon-Mediated Missplicing of SMOC2. Curr. Biol. 2017, 27, 1573–1584.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Performagene. Laboratory Protocol for Manual Purification of DNA from 0.5 mL of Performagene™ Sample. Available online: https://www.dnagenotek.com/US/pdf/PD-PR-083.pdf (accessed on 30 August 2021).
- Desjardins, P.; Conklin, D. NanoDrop Microvolume Quantitation of Nucleic Acids. J. Vis. Exp. 2010, 45, e2565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scientific, T. User Guide: Qubit dsDNA HS Assay Kits. Available online: https://www.thermofisher.com/document-connect/document-connect.html?url=https%3A%2F%2Fassets.thermofisher.com%2FTFS-Assets%2FLSG%2Fmanuals%2FQubit_dsDNA_HS_Assay_UG.pdf&title=VXNlciBHdWlkZTogUXViaXQgZHNETkEgSFMgQXNzYXkgS2l0cw== (accessed on 20 August 2021).
- Scientific, T. Quant-iT PicoGreen dsDNA Reagent and Kits. Available online: https://www.thermofisher.com/document-connect/document-connect.html?url=https%3A%2F%2Fassets.thermofisher.com%2FTFS-Assets%2FLSG%2Fmanuals%2Fmp07581.pdf&title=UXVhbnQtaVQgUGljb0dyZWVuIGRzRE5BIFJlYWdlbnQgYW5kIEtpdHM= (accessed on 20 August 2021).
- Van Der Auwera, G.A.; Carneiro, M.O.; Hartl, C.; Poplin, R.; Del Angel, G.; Levy-Moonshine, A.; Jordan, T.; Shakir, K.; Roazen, D.; Thibault, J.; et al. From FastQ Data to High-Confidence Variant Calls: The Genome Analysis Toolkit Best Practices Pipeline. Curr. Protoc. Bioinform. 2013, 43, 11.10.1–11.10.33. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- Broad Institute Picard Tools. 2016. Available online: https://broadinstitute.github.io/picard/ (accessed on 12 July 2021).
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; dePristo, M.A.; Handsaker, R.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef] [PubMed]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.; Daly, M.J.; et al. PLINK: A Tool Set for Whole-Genome Association and Population-Based Linkage Analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [Green Version]
- Chang, C.; Chow, C.C.; Tellier, L.C.A.M.; Vattikuti, S.; Purcell, S.M.; Lee, J.J. Second-generation PLINK: Rising to the challenge of larger and richer datasets. GigaScience 2015, 4, 7. [Google Scholar] [CrossRef]
- Shaun Purcell, C.C. PLINK version 1.9b. Available online: www.cog-genomics.org/plink/1.9/ (accessed on 20 August 2021).
- Wickham, H. Ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016; ISBN 978-3-319-24277-4. Available online: https://ggplot2.tidyverse.org (accessed on 20 August 2021).
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing; R Core Team: Vienna, Austria, 2021. [Google Scholar]
- R Studio Team. R Studio: Integrated Development for R; R Studio: Boston, MA, USA, 2020. [Google Scholar]
- Raj, A.; Stephens, M.; Pritchard, J.K. fastSTRUCTURE: Variational Inference of Population Structure in Large SNP Data Sets. Genetics 2014, 197, 573–589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wickham, H.; Francois, R.; Henry, L.; Müller, K. dplyr: A Grammar of Data Manipulation. R Package Version 0.4.3. Available online: https://CRAN.R-project.org/package=dplyr (accessed on 20 August 2021).
- Kopelman, N.M.; Mayzel, J.; Jakobsson, M.; Rosenberg, N.; Mayrose, I. Clumpak: A program for identifying clustering modes and packaging population structure inferences across K. Mol. Ecol. Resour. 2015, 15, 1179–1191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Francis, R.M. pophelper: An R package and web app to analyse and visualize population structure. Mol. Ecol. Resour. 2016, 17, 27–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertrand, A.R.; Kadri, N.K.; Flori, L.; Gautier, M.; Druet, T. RZooRoH: An R package to characterize individual genomic autozygosity and identify homozygous-by-descent segments. Methods Ecol. Evol. 2019, 10, 860–866. [Google Scholar] [CrossRef]
- GCCF. Asian. Available online: https://www.gccfcats.org/Cat-Breeds/Asian (accessed on 27 August 2021).
- GCCF. Oriental. Available online: https://www.gccfcats.org/Cat-Breeds/Oriental (accessed on 20 August 2021).
- GCCF. GCCF Tonkinese Registration Policy. Available online: https://www.gccfcats.org/Portals/0/Tonkanese.RegPol.Feb18.pdf?ver=2018-02-25-092455-197 (accessed on 20 August 2021).
- GCCF. Siberian. Available online: https://www.gccfcats.org/Cat-Breeds/Siberian (accessed on 20 August 2021).
- Canine and Feline Sector Group. Breeding and Sale of cats report 2015. Available online: https://www.cfsg.org.uk/repository/247/ (accessed on 20 August 2021).
- Pearce, W. Ragdoll History-The Beginning | Ragdoll Historical Society. Available online: http://ragdollhistoricalsociety.org/ragdoll-history-the-beginning/ (accessed on 26 March 2021).
- TICA. Maine Coon Breed. Available online: https://tica.org/breeds/browse-all-breeds?view=article&id=853:maine-coon-breed&catid=79 (accessed on 6 March 2021).
- Taylor, D.; Negus, D. The Ultimate Cat Book; Simon and Schuster: New York, NY, USA, 1989. [Google Scholar]
- Boyko, A.R.; Boyko, R.H.; Boyko, C.M.; Parker, H.G.; Castelhano, M.; Corey, L.; Degenhardt, J.D.; Auton, A.; Hedimbi, M.; Kityo, R.; et al. Complex population structure in African village dogs and its implications for inferring dog domestication history. Proc. Natl. Acad. Sci. USA 2009, 106, 13903–13908. [Google Scholar] [CrossRef] [Green Version]
- Faure, E.; Kitchener, A.C. An archaeological and historical review of the relationships between felids and people. Anthrozoös 2009, 22, 221–238. [Google Scholar] [CrossRef]
- Davis, S.J. The Archaeology of Animals; B.T. Batsford Ltd.: London, UK, 1987. [Google Scholar]
- Harcourt, R.A. The animal bones. In Gussange All Saints: An Iron Age Settlement in Dorset; Department of the Environment Archaeological Reports 10; Her Majesty’s Stationary Office: London, UK, 1979; pp. 150–160. [Google Scholar]
- Yahara, T.; Kasuya, E.; Yamane, A.; Ishida, Y. Female control of paternity during copulation: Inbreeding avoidance in feral cats. Behaviour 2001, 138, 235–250. [Google Scholar] [CrossRef]
- Mucha, S.; Wolc, A.; Gradowska, A.; Szwaczkowski, T. Inbreeding rate and genetic structure of cat populations in Poland. J. Appl. Genet. 2010, 52, 101–110. [Google Scholar] [CrossRef] [Green Version]
- Albrechtsen, A.; Nielsen, F.C.; Nielsen, R. Ascertainment Biases in SNP Chips Affect Measures of Population Divergence. Mol. Biol. Evol. 2010, 27, 2534–2547. [Google Scholar] [CrossRef] [Green Version]
- Sutter, N.B.; Eberle, M.; Parker, H.G.; Pullar, B.J.; Kirkness, E.F.; Kruglyak, L.; Ostrander, E. Extensive and breed-specific linkage disequilibrium in Canis familiaris. Genome Res. 2004, 14, 2388–2396. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Quality Control Process | Genotyping Rate | Variants | Samples |
|---|---|---|---|
| Raw genotypes | 0.95 | 263,482 | 1344 |
| Remove duplicate samples: 34 cats | 0.95 | 263,482 | 1310 |
| 1st pass remove variants (--geno 0.2): 14,257 SNVs | 0.95 | 249,225 | 1310 |
| 1st pass remove samples (--mind 0.2): 9 cats | 0.99 | 249,225 | 1301 |
| 2nd pass remove variants (--geno 0.02): 32,427 SNVs | 0.99 | 216,798 | 1301 |
| 2nd pass remove samples (--mind 0.05): 11 cats | 0.99 | 216,798 | 1290 |
| MAF <= 0.05 (--maf 0.05): 38,292 SNVs removed | 0.99 | 178,506 | 1290 |
| Analysis-ready genotype callset | 0.99 | 178,506 | 1290 |
| Breed | Number of Samples | Accepted Date of Origin | GCCF 2020 | MAF | Ho | FIS | Monomorphic SNVs | Informative SNzVs | Area of Origin |
|---|---|---|---|---|---|---|---|---|---|
| Southeast Asia | |||||||||
| Balinese | 2 | 1940s | 26 | 0.27 | 0.21 | −0.216 | 61.71% | 38.29% | USA |
| Burmese | 27 | 1350–1767 | 1091 | 0.27 | 0.19 | 0.073 | 24.93% | 75.07% | Thailand |
| Korat | 1 | 1350–1767 | 42 | 0.25 | 0.18 | N/A | 82.05% | 17.95% | Thailand |
| Oriental | 6 | 1950s | 619 1 | 0.27 | 0.20 | −0.035 | 45.08% | 54.92% | UK |
| Siamese | 36 | 1350–1767 | 1631 | 0.27 | 0.20 | 0.088 | 19.83% | 80.17% | Thailand |
| Tonkinese | 10 | 1950s | 169 | 0.27 | 0.23 | −0.042 | 32.28% | 67.72% | USA |
| Mediterranean | |||||||||
| Abyssinian | 7 | 1860s | 143 | 0.25 | 0.24 | −0.017 | 32.66% | 67.34% | Ethiopia |
| Egyptian Mau | 2 | Early | 58 | 0.25 | 0.25 | −0.06 | 45.93% | 54.07% | Egypt |
| Somali | 2 | 1967 | 981 | 0.25 | 0.24 | −0.211 | 56.54% | 43.46% | USA/Canada |
| Turkish angora | 1 | Early | N/A | 0.26 | 0.30 | N/A | 69.68% | 30.32% | Turkey |
| Turkish van | 2 | Early | 8 | 0.25 | 0.22 | −0.009 | 48.06% | 51.94% | Turkey |
| Western | |||||||||
| American shorthair | 1 | 1900 | N/A | 0.25 | 0.32 | N/A | 67.94% | 32.06% | USA |
| Cornish rex | 2 | 1950s | 71 | 0.26 | 0.27 | −0.208 | 49.00% | 51.00% | UK |
| Devon rex | 8 | 1960s | 321 | 0.25 | 0.26 | −0.04 | 29.47% | 70.53% | UK |
| Domestic longhair | 74 | N/A | N/A | 0.25 | 0.29 | 0.077 | 10.05% | 89.95% | UK |
| Domestic semi-longhair | 12 | N/A | N/A | 0.25 | 0.28 | 0.066 | 16.34% | 83.66% | UK |
| Domestic shorthair | 754 | N/A | N/A | 0.24 | 0.30 | 0.058 | 8.17% | 91.83% | UK |
| Maine coon | 47 | 1860s | 2566 | 0.25 | 0.27 | 0.061 | 13.66% | 86.34% | USA |
| Manx | 2 | Early | 21 | 0.25 | 0.28 | N/A | 44.88% | 55.12% | UK |
| Norwegian forest cat | 15 | Early | 301 | 0.25 | 0.30 | −0.001 | 17.24% | 82.76% | Norway |
| Siberian | 6 | Early | 358 | 0.25 | 0.28 | −0.001 | 24.53% | 75.47% | Russia |
| Sphynx | 8 | 1966 | 196 | 0.25 | 0.27 | −0.04 | 27.23% | 72.77% | Canada |
| Tiffany | 1 | 1967 | N/A | 0.26 | 0.24 | N/A | 75.58% | 24.42% | USA |
| Persian | |||||||||
| Persian 2 | 51 | Early | 908 | 0.24 | 0.25 | 0.113 | 14.77% | 85.23% | Iran |
| Persian/Western | |||||||||
| British shorthair | 95 | 1870’s | 9111 | 0.24 | 0.26 | 0.096 | 11.70% | 88.30% | UK |
| Exotic shorthair | 15 | 1966 | 350 | 0.25 | 0.25 | −0.017 | 29.09% | 70.91% | USA |
| Southeast Asia/Persian | |||||||||
| Asian | 2 | 1981 | 187 3 | 0.26 | 0.20 | −0.163 | 61.35% | 38.65% | UK |
| Birman | 10 | 1930s | 472 | 0.26 | 0.21 | 0.024 | 30.24% | 69.76% | Burma |
| Hybrid | |||||||||
| Bengal | 27 | 1963 | 263 | 0.26 | 0.25 | 0.082 | 16.96% | 83.04% | USA |
| Savannah cat | 1 | 1997 | N/A | 0.26 | 0.28 | N/A | 71.83% | 28.17% | UK |
| Eastern/Western | |||||||||
| American bobtail | 1 | 1960s | N/A | 0.25 | 0.32 | N/A | 68.29% | 31.71% | USA |
| Bombay | 1 | 1958 | N/A 4 | N/A | 0.20 | −0.5 | 79.73% | 20.27% | USA |
| Ragdoll | 27 | 1960s | 4387 | 0.25 | 0.24 | 0.056 | 19.26% | 80.74% | USA |
| Russian blue | 6 | Late 1800s | 443 | 0.25 | 0.25 | −0.071 | 35.77% | 64.23% | Russia |
| Selkirk rex | 1 | 1987 | 89 | 0.24 | 0.26 | N/A | 73.55% | 26.45% | USA |
| Snowshoe | 2 | 1960s | 73 | 0.26 | 0.29 | −0.16 | 44.85% | 55.15% | USA |
| Unknown | |||||||||
| Cross | 17 | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A |
| Unknown | 8 | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A |
| Total | 1290 | ||||||||
| Breed | Number | MAF | Ho | FIS | Monomorphic SNVs | Informative SNVs |
|---|---|---|---|---|---|---|
| Abyssinian | 7 | 0.25 | 0.24 | −0.017 | 32.66% | 67.34% |
| Bengal | 10 | 0.25 | 0.25 | −0.020 | 29.17% | 70.83% |
| Birman | 10 | 0.23 | 0.21 | 0.024 | 30.24% | 69.76% |
| British shorthair | 10 | 0.25 | 0.27 | 0.030 | 21.07% | 78.93% |
| Burmese | 10 | 0.23 | 0.20 | 0.031 | 32.22% | 67.78% |
| Devon rex | 8 | 0.24 | 0.26 | −0.040 | 29.47% | 70.53% |
| Domestic longhair | 10 | 0.26 | 0.30 | 0.006 | 17.62% | 82.38% |
| Domestic shorthair | 10 | 0.26 | 0.31 | −0.036 | 17.85% | 82.15% |
| Domestic semi-longhair | 10 | 0.26 | 0.28 | 0.059 | 17.55% | 82.45% |
| Exotic | 9 | 0.25 | 0.25 | 0.017 | 29.09% | 70.91% |
| Maine coon | 10 | 0.25 | 0.26 | 0.082 | 20.54% | 79.46% |
| Norwegian forest cat | 10 | 0.26 | 0.30 | −0.009 | 19.41% | 80.59% |
| Oriental | 6 | 0.23 | 0.20 | −0.035 | 45.08% | 54.92% |
| Persian | 10 | 0.25 | 0.25 | 0.108 | 22.28% | 77.72% |
| Ragdoll | 10 | 0.24 | 0.23 | 0.005 | 31.63% | 68.37% |
| Russian blue | 6 | 0.25 | 0.25 | −0.071 | 35.77% | 64.23% |
| Siamese | 10 | 0.22 | 0.19 | 0.026 | 38.07% | 61.93% |
| Siberian | 6 | 0.25 | 0.28 | −0.001 | 24.53% | 75.47% |
| Sphynx | 8 | 0.25 | 0.27 | −0.040 | 27.23% | 72.77% |
| Tonkinese | 10 | 0.23 | 0.23 | −0.042 | 32.28% | 67.72% |
| Total | 180 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Irving McGrath, J.; Zhang, W.; Hollar, R.; Collings, A.; Powell, R.; Foale, R.D.; Thurley, N.; Brockman, J.A.; Mellanby, R.J.; Gunn-Moore, D.A.; et al. More Than a Moggy; A Population Genetics Analysis of the United Kingdom’s Non-Pedigree Cats. Genes 2021, 12, 1619. https://doi.org/10.3390/genes12101619
Irving McGrath J, Zhang W, Hollar R, Collings A, Powell R, Foale RD, Thurley N, Brockman JA, Mellanby RJ, Gunn-Moore DA, et al. More Than a Moggy; A Population Genetics Analysis of the United Kingdom’s Non-Pedigree Cats. Genes. 2021; 12(10):1619. https://doi.org/10.3390/genes12101619
Chicago/Turabian StyleIrving McGrath, Jennifer, Wengang Zhang, Regina Hollar, Alison Collings, Roger Powell, Rob D. Foale, Nicola Thurley, Jeffrey A. Brockman, Richard J. Mellanby, Danièlle A. Gunn-Moore, and et al. 2021. "More Than a Moggy; A Population Genetics Analysis of the United Kingdom’s Non-Pedigree Cats" Genes 12, no. 10: 1619. https://doi.org/10.3390/genes12101619