Inhibition of the Lipid Droplet–Peroxisome Proliferator-Activated Receptor α Axis Suppresses Cancer Stem Cell Properties

 ,
,
Abstract
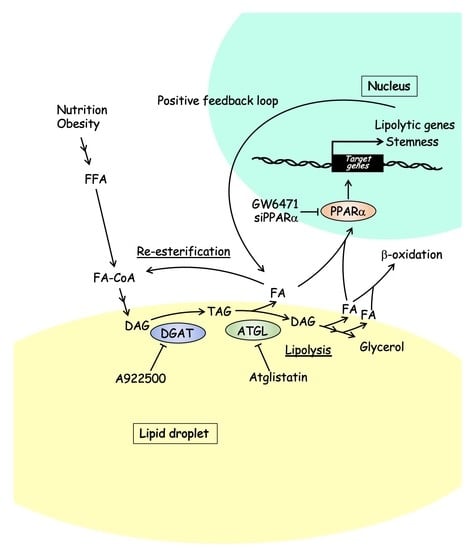
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Reagents and Antibodies
2.2. Cell Culture
2.3. Gene Silencing by siRNA
2.4. Immunoblot Analysis
2.5. LD Staining
2.6. Reverse Transcription-PCR (RT-PCR)
2.7. Sphere Formation Analysis
2.8. TAG Measurement Analysis
2.9. Statistical Analysis
3. Results
3.1. LDs Develop More in CSCs Than in Non-CSCs
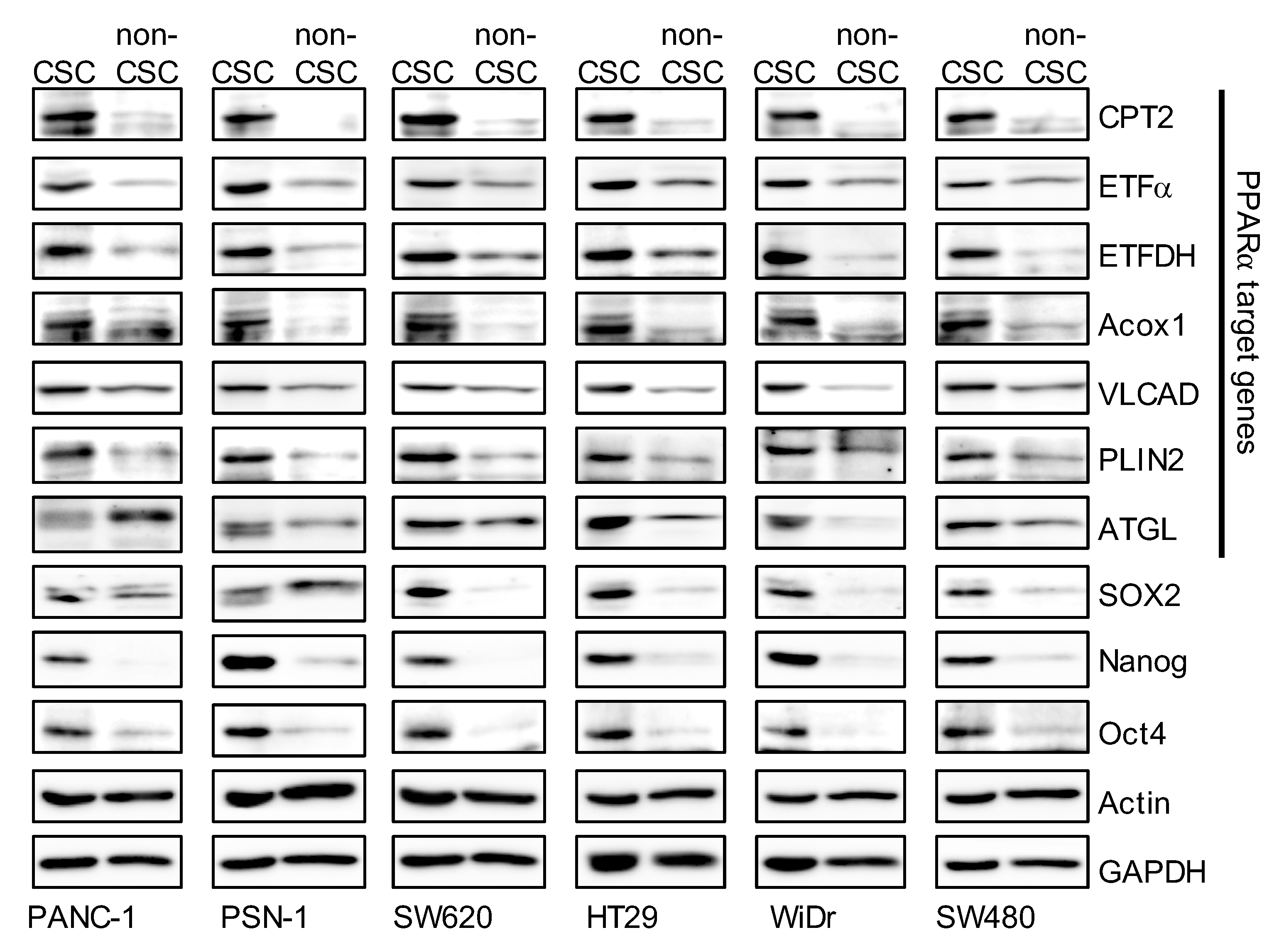
3.2. The Peroxisome Proliferator-Activated Receptor α (PPARα) Pathway is Activated in CSCs
3.3. Inhibition of the PPARα Suppresses the Expression of CSC Markers
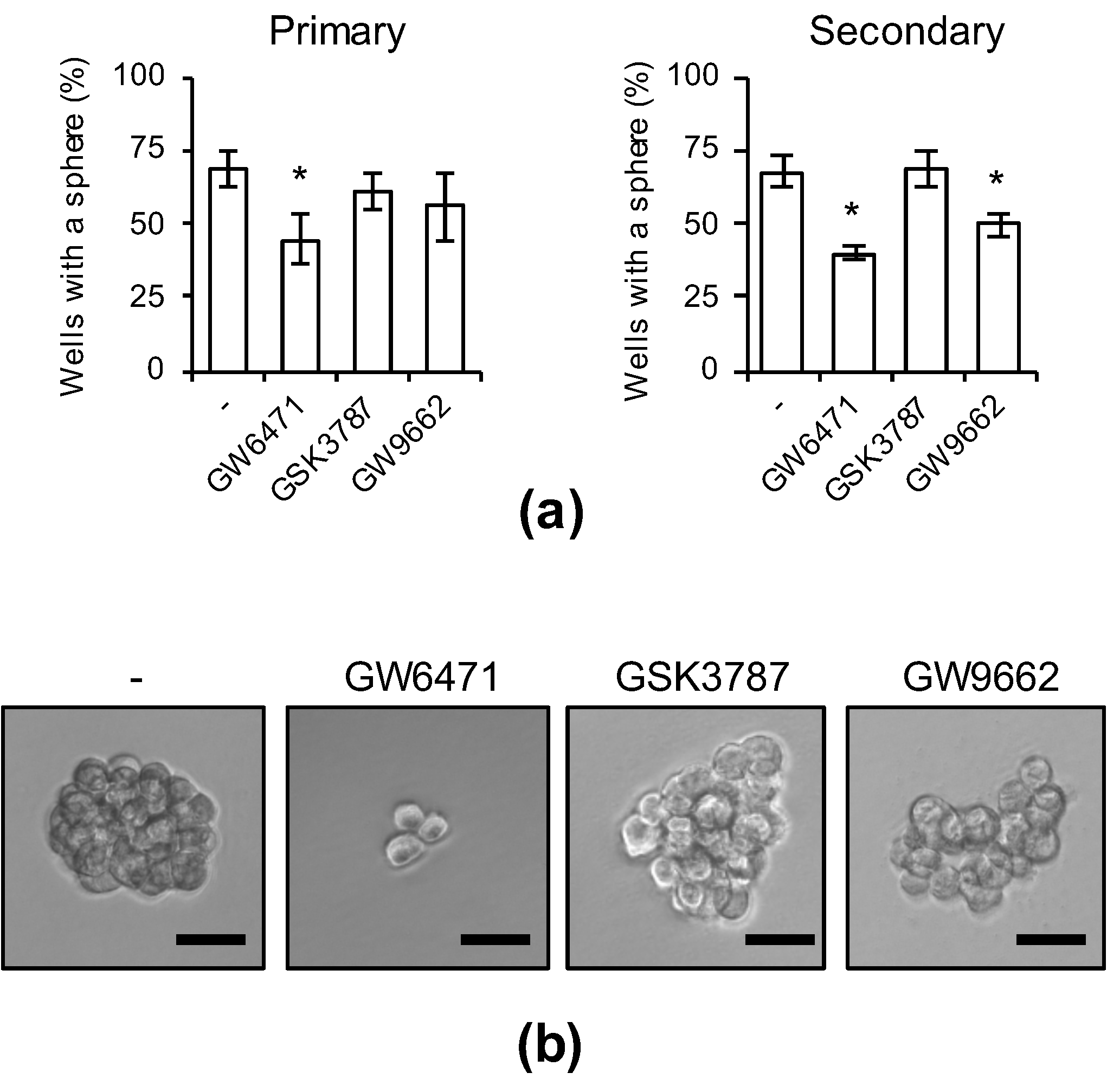
3.4. PPARα Suppression Inhibits the Sphere-Formation Ability of CSCs
3.5. Genetic Inhibition of PPARα Suppresses Cancer Stemness
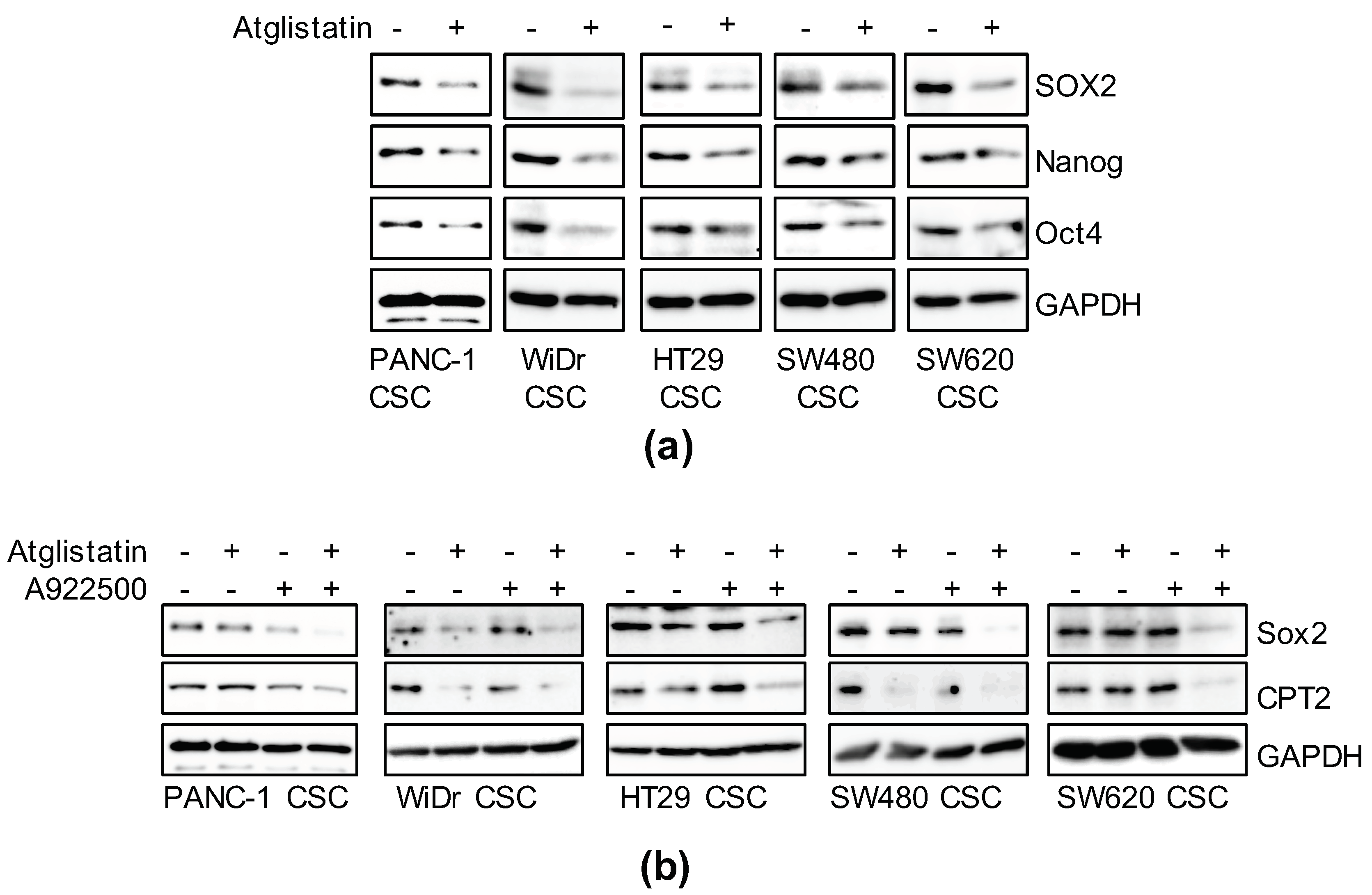
3.6. Inhibition of TAG Turnover Suppresses Cancer Stemness
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sancho, P.; Burgos-Ramos, E.; Tavera, A.; Bou Kheir, T.; Jagust, P.; Schoenhals, M.; Barneda, D.; Sellers, K.; Campos-Olivas, R.; Graña, O.; et al. MYC/PGC-1α Balance Determines the Metabolic Phenotype and Plasticity of Pancreatic Cancer Stem Cells. Cell Metab. 2015, 22, 590–605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lagadinou, E.D.; Sach, A.; Callahan, K.; Rossi, R.M.; Neering, S.J.; Minhajuddin, M.; Ashton, J.M.; Pei, S.; Grose, V.; O’Dwyer, K.M.; et al. BCL-2 inhibition targets oxidative phosphorylation and selectively eradicates quiescent human leukemia stem cells. Cell Stem Cell 2013, 12, 329–341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janiszewska, M.; Suvà, M.L.; Riggi, N.; Houtkooper, R.H.; Auwerx, J.; Clément-Schatlo, V.; Radovanovic, I.; Rheinbay, E.; Provero, P.; Stamenkovic, I. Imp2 controls oxidative phosphorylation and is crucial for preserving glioblastoma cancer stem cells. Genes Dev. 2012, 26, 1926–1944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Viale, A.; Pettazzoni, P.; Lyssiotis, C.A.; Ying, H.; Sánchez, N.; Marchesini, M.; Carugo, A.; Green, T.; Seth, S.; Giuliani, V.; et al. Oncogene ablation-resistant pancreatic cancer cells depend on mitochondrial function. Nature 2014, 514, 628–632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pastò, A.; Bellio, C.; Pilotto, G.; Ciminale, V.; Silic-Benussi, M.; Guzzo, G.; Rasola, A.; Frasson, C.; Nardo, G.; Zulato, E.; et al. Cancer stem cells from epithelial ovarian cancer patients privilege oxidative phosphorylation, and resist glucose deprivation. Oncotarget 2014, 5, 4305–4319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vlashi, E.; Lagadec, C.; Vergnes, L.; Reue, K.; Frohnen, P.; Chan, M.; Alhiyari, Y.; Dratver, M.B.; Pajonk, F. Metabolic differences in breast cancer stem cells and differentiated progeny. Breast Cancer Res. Treat. 2014, 146, 525–534. [Google Scholar] [CrossRef] [Green Version]
- Kuramoto, K.; Yamamoto, M.; Suzuki, S.; Sanomachi, T.; Togashi, K.; Seino, S.; Kitanaka, C.; Okada, M. Verteporfin inhibits oxidative phosphorylation and induces cell death specifically in glioma stem cells. FEBS J. 2020, 287, 2023–2036. [Google Scholar] [CrossRef]
- Kuramoto, K.; Suzuki, S.; Sakaki, H.; Takeda, H.; Sanomachi, T.; Seino, S.; Narita, Y.; Kayama, T.; Kitanaka, C.; Okada, M. Licochalcone A specifically induces cell death in glioma stem cells via mitochondrial dysfunction. FEBS Open Bio 2017, 7, 835–844. [Google Scholar] [CrossRef]
- Fiorillo, M.; Lamb, R.; Tanowitz, H.B.; Mutti, L.; Krstic-Demonacos, M.; Cappello, A.R.; Martinez-Outschoorn, U.E.; Sotgia, F.; Lisanti, M.P. Repurposing atovaquone: Targeting mitochondrial complex III and OXPHOS to eradicate cancer stem cells. Oncotarget 2016, 7, 34084–34099. [Google Scholar] [CrossRef] [Green Version]
- Cruz, A.L.S.; Barreto, E.A.; Fazolini, N.P.B.; Viola, J.P.B.; Bozza, P.T. Lipid droplets: Platforms with multiple functions in cancer hallmarks. Cell Death Dis. 2020, 11, 105. [Google Scholar] [CrossRef] [Green Version]
- Hager, M.H.; Solomon, K.R.; Freeman, M.R. The role of cholesterol in prostate cancer. Curr. Opin. Clin. Nutr. Metab. Care 2006, 9, 379–385. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y. Fatty acid oxidation is a dominant bioenergetic pathway in prostate cancer. Prostate Cancer Prostatic Dis. 2006, 9, 230–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walther, T.C.; Farese, R.V., Jr. Lipid droplets and cellular lipid metabolism. Annu. Rev. Biochem. 2012, 81, 687–714. [Google Scholar] [CrossRef] [Green Version]
- Zechner, R.; Zimmermann, R.; Eichmann, T.O.; Kohlwein, S.D.; Haemmerle, G.; Lass, A.; Madeo, F. FAT SIGNALS—Lipases and lipolysis in lipid metabolism and signaling. Cell Metab. 2012, 15, 279–291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krahmer, N.; Farese, R.V., Jr.; Walther, T.C. Balancing the fat: Lipid droplets and human disease. EMBO Mol. Med. 2013, 5, 973–983. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, A.S.; Coleman, R.A.; Kraemer, F.B.; McManaman, J.L.; Obin, M.S.; Puri, V.; Yan, Q.W.; Miyoshi, H.; Mashek, D.G. The role of lipid droplets in metabolic disease in rodents and humans. J. Clin. Investig. 2011, 121, 2102–2110. [Google Scholar] [CrossRef] [Green Version]
- Arrese, E.L.; Saudale, F.Z.; Soulages, J.L. Lipid Droplets as Signaling Platforms Linking Metabolic and Cellular Functions. Lipid Insights 2014, 7, 7–16. [Google Scholar] [CrossRef]
- Li, J.; Condello, S.; Thomes-Pepin, J.; Ma, X.; Xia, Y.; Hurley, T.D.; Matei, D.; Cheng, J.X. Lipid Desaturation Is a Metabolic Marker and Therapeutic Target of Ovarian Cancer Stem Cells. Cell Stem Cell 2017, 20, 303–314.e305. [Google Scholar] [CrossRef] [Green Version]
- Hoang-Minh, L.B.; Siebzehnrubl, F.A.; Yang, C.; Suzuki-Hatano, S.; Dajac, K.; Loche, T.; Andrews, N.; Schmoll Massari, M.; Patel, J.; Amin, K.; et al. Infiltrative and drug-resistant slow-cycling cells support metabolic heterogeneity in glioblastoma. EMBO J. 2018, 37. [Google Scholar] [CrossRef]
- Tirinato, L.; Liberale, C.; Di Franco, S.; Candeloro, P.; Benfante, A.; La Rocca, R.; Potze, L.; Marotta, R.; Ruffilli, R.; Rajamanickam, V.P.; et al. Lipid droplets: A new player in colorectal cancer stem cells unveiled by spectroscopic imaging. Stem Cells 2015, 33, 35–44. [Google Scholar] [CrossRef] [Green Version]
- Visweswaran, M.; Arfuso, F.; Warrier, S.; Dharmarajan, A. Aberrant lipid metabolism as an emerging therapeutic strategy to target cancer stem cells. Stem Cells 2020, 38, 6–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tirinato, L.; Pagliari, F.; Limongi, T.; Marini, M.; Falqui, A.; Seco, J.; Candeloro, P.; Liberale, C.; Di Fabrizio, E. An Overview of Lipid Droplets in Cancer and Cancer Stem Cells. Stem Cells Int. 2017, 2017, 1656053. [Google Scholar] [CrossRef]
- Singh, S.R.; Zeng, X.; Zhao, J.; Liu, Y.; Hou, G.; Liu, H.; Hou, S.X. The lipolysis pathway sustains normal and transformed stem cells in adult Drosophila. Nature 2016, 538, 109–113. [Google Scholar] [CrossRef] [PubMed]
- Haemmerle, G.; Moustafa, T.; Woelkart, G.; Büttner, S.; Schmidt, A.; Van de Weijer, T.; Hesselink, M.; Jaeger, D.; Kienesberger, P.C.; Zierler, K.; et al. ATGL-mediated fat catabolism regulates cardiac mitochondrial function via PPAR-α and PGC-1. Nat. Med. 2011, 17, 1076–1085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furuta, S.; Miyazawa, S.; Hashimoto, T. Purification and properties of rat liver acyl-CoA dehydrogenases and electron transfer flavoprotein. J. Biochem. 1981, 90, 1739–1750. [Google Scholar] [CrossRef]
- Miyazawa, S.; Ozasa, H.; Osumi, T.; Hashimoto, T. Purification and properties of carnitine octanoyltransferase and carnitine palmitoyltransferase from rat liver. J. Biochem. 1983, 94, 529–542. [Google Scholar] [CrossRef]
- Kuramoto, K.; Sakai, F.; Yoshinori, N.; Nakamura, T.Y.; Wakabayashi, S.; Kojidani, T.; Haraguchi, T.; Hirose, F.; Osumi, T. Deficiency of a lipid droplet protein, perilipin 5, suppresses myocardial lipid accumulation, thereby preventing type 1 diabetes-induced heart malfunction. Mol. Cell Biol. 2014, 34, 2721–2731. [Google Scholar] [CrossRef] [Green Version]
- Okada, M.; Shibuya, K.; Sato, A.; Seino, S.; Suzuki, S.; Seino, M.; Kitanaka, C. Targeting the K-Ras--JNK axis eliminates cancer stem-like cells and prevents pancreatic tumor formation. Oncotarget 2014, 5, 5100–5112. [Google Scholar] [CrossRef] [Green Version]
- Pollard, S.M.; Yoshikawa, K.; Clarke, I.D.; Danovi, D.; Stricker, S.; Russell, R.; Bayani, J.; Head, R.; Lee, M.; Bernstein, M.; et al. Glioma stem cell lines expanded in adherent culture have tumor-specific phenotypes and are suitable for chemical and genetic screens. Cell Stem Cell 2009, 4, 568–580. [Google Scholar] [CrossRef] [Green Version]
- Okada, M.; Takeda, H.; Sakaki, H.; Kuramoto, K.; Suzuki, S.; Sanomachi, T.; Togashi, K.; Seino, S.; Kitanaka, C. Repositioning CEP-1347, a chemical agent originally developed for the treatment of Parkinson’s disease, as an anti-cancer stem cell drug. Oncotarget 2017, 8, 94872–94882. [Google Scholar] [CrossRef] [Green Version]
- Sato, A.; Okada, M.; Shibuya, K.; Watanabe, E.; Seino, S.; Suzuki, K.; Narita, Y.; Shibui, S.; Kayama, T.; Kitanaka, C. Resveratrol promotes proteasome-dependent degradation of Nanog via p53 activation and induces differentiation of glioma stem cells. Stem Cell Res. 2013, 11, 601–610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rakhshandehroo, M.; Knoch, B.; Müller, M.; Kersten, S. Peroxisome proliferator-activated receptor α target genes. PPAR Res. 2010, 2010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vegliante, R.; Di Leo, L.; Ciccarone, F.; Ciriolo, M.R. Hints on ATGL implications in cancer: Beyond bioenergetic clues. Cell Death Dis. 2018, 9, 316. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, R.; Strauss, J.G.; Haemmerle, G.; Schoiswohl, G.; Birner-Gruenberger, R.; Riederer, M.; Lass, A.; Neuberger, G.; Eisenhaber, F.; Hermetter, A.; et al. Fat mobilization in adipose tissue is promoted by adipose triglyceride lipase. Science 2004, 306, 1383–1386. [Google Scholar] [CrossRef] [Green Version]
- Mayer, N.; Schweiger, M.; Romauch, M.; Grabner, G.F.; Eichmann, T.O.; Fuchs, E.; Ivkovic, J.; Heier, C.; Mrak, I.; Lass, A.; et al. Development of small-molecule inhibitors targeting adipose triglyceride lipase. Nat. Chem. Biol. 2013, 9, 785–787. [Google Scholar] [CrossRef] [Green Version]
- Sathyanarayan, A.; Mashek, M.T.; Mashek, D.G. ATGL Promotes Autophagy/Lipophagy via SIRT1 to Control Hepatic Lipid Droplet Catabolism. Cell Rep. 2017, 19, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Han, S.; Hu, L.; Quach, T.; Simpson, J.S.; Trevaskis, N.L.; Porter, C.J. Profiling the role of deacylation-reacylation in the lymphatic transport of a triglyceride-mimetic prodrug. Pharm. Res. 2015, 32, 1830–1844. [Google Scholar] [CrossRef]
- Chen, H.C.; Farese, R.V., Jr. DGAT and triglyceride synthesis: A new target for obesity treatment? Trends Cardiovasc. Med. 2000, 10, 188–192. [Google Scholar] [CrossRef]
- Ma, X.L.; Sun, Y.F.; Wang, B.L.; Shen, M.N.; Zhou, Y.; Chen, J.W.; Hu, B.; Gong, Z.J.; Zhang, X.; Cao, Y.; et al. Sphere-forming culture enriches liver cancer stem cells and reveals Stearoyl-CoA desaturase 1 as a potential therapeutic target. BMC Cancer 2019, 19, 760. [Google Scholar] [CrossRef] [Green Version]
- Haynes, H.R.; White, P.; Hares, K.M.; Redondo, J.; Kemp, K.C.; Singleton, W.G.B.; Killick-Cole, C.L.; Stevens, J.R.; Garadi, K.; Guglani, S.; et al. The transcription factor PPARα is overexpressed and is associated with a favourable prognosis in IDH-wildtype primary glioblastoma. Histopathology 2017, 70, 1030–1043. [Google Scholar] [CrossRef]
- Haynes, H.R.; Scott, H.L.; Killick-Cole, C.L.; Shaw, G.; Brend, T.; Hares, K.M.; Redondo, J.; Kemp, K.C.; Ballesteros, L.S.; Herman, A.; et al. shRNA-mediated PPARα knockdown in human glioma stem cells reduces in vitro proliferation and inhibits orthotopic xenograft tumour growth. J. Pathol. 2019, 247, 422–434. [Google Scholar] [CrossRef] [PubMed]
- Grabacka, M.; Pierzchalska, M.; Reiss, K. Peroxisome proliferator activated receptor α ligands as anticancer drugs targeting mitochondrial metabolism. Curr. Pharm. Biotechnol. 2013, 14, 342–356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilk, A.; Wyczechowska, D.; Zapata, A.; Dean, M.; Mullinax, J.; Marrero, L.; Parsons, C.; Peruzzi, F.; Culicchia, F.; Ochoa, A.; et al. Molecular mechanisms of fenofibrate-induced metabolic catastrophe and glioblastoma cell death. Mol. Cell Biol. 2015, 35, 182–198. [Google Scholar] [CrossRef] [Green Version]
- Binello, E.; Mormone, E.; Emdad, L.; Kothari, H.; Germano, I.M. Characterization of fenofibrate-mediated anti-proliferative pro-apoptotic effects on high-grade gliomas and anti-invasive effects on glioma stem cells. J. Neurooncol. 2014, 117, 225–234. [Google Scholar] [CrossRef] [PubMed]
- Stebbins, K.J.; Broadhead, A.R.; Cabrera, G.; Correa, L.D.; Messmer, D.; Bundey, R.; Baccei, C.; Bravo, Y.; Chen, A.; Stock, N.S.; et al. In vitro and in vivo pharmacology of NXT629, a novel and selective PPARα antagonist. Eur. J. Pharm. 2017, 809, 130–140. [Google Scholar] [CrossRef]
- Messmer, D.; Lorrain, K.; Stebbins, K.; Bravo, Y.; Stock, N.; Cabrera, G.; Correa, L.; Chen, A.; Jacintho, J.; Chiorazzi, N.; et al. A Selective Novel Peroxisome Proliferator-Activated Receptor (PPAR)-α Antagonist Induces Apoptosis and Inhibits Proliferation of CLL Cells In Vitro and In Vivo. Mol. Med. 2015, 21, 410–419. [Google Scholar] [CrossRef]
- Han, S.; Wei, R.; Zhang, X.; Jiang, N.; Fan, M.; Huang, J.H.; Xie, B.; Zhang, L.; Miao, W.; Butler, A.C.; et al. CPT1A/2-Mediated FAO Enhancement-A Metabolic Target in Radioresistant Breast Cancer. Front. Oncol. 2019, 9, 1201. [Google Scholar] [CrossRef]
- Ito, K.; Carracedo, A.; Weiss, D.; Arai, F.; Ala, U.; Avigan, D.E.; Schafer, Z.T.; Evans, R.M.; Suda, T.; Lee, C.H.; et al. A PML–PPAR-δ pathway for fatty acid oxidation regulates hematopoietic stem cell maintenance. Nat. Med. 2012, 18, 1350–1358. [Google Scholar] [CrossRef] [Green Version]
- Prost, S.; Relouzat, F.; Spentchian, M.; Ouzegdouh, Y.; Saliba, J.; Massonnet, G.; Beressi, J.P.; Verhoeyen, E.; Raggueneau, V.; Maneglier, B.; et al. Erosion of the chronic myeloid leukaemia stem cell pool by PPARγ agonists. Nature 2015, 525, 380–383. [Google Scholar] [CrossRef]
- Rousselot, P.; Prost, S.; Guilhot, J.; Roy, L.; Etienne, G.; Legros, L.; Charbonnier, A.; Coiteux, V.; Cony-Makhoul, P.; Huguet, F.; et al. Pioglitazone together with imatinib in chronic myeloid leukemia: A proof of concept study. Cancer 2017, 123, 1791–1799. [Google Scholar] [CrossRef]
- Villena, J.A.; Roy, S.; Sarkadi-Nagy, E.; Kim, K.H.; Sul, H.S. Desnutrin, an adipocyte gene encoding a novel patatin domain-containing protein, is induced by fasting and glucocorticoids: Ectopic expression of desnutrin increases triglyceride hydrolysis. J. Biol. Chem. 2004, 279, 47066–47075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshii, Y.; Furukawa, T.; Yoshii, H.; Mori, T.; Kiyono, Y.; Waki, A.; Kobayashi, M.; Tsujikawa, T.; Kudo, T.; Okazawa, H.; et al. Cytosolic acetyl-CoA synthetase affected tumor cell survival under hypoxia: The possible function in tumor acetyl-CoA/acetate metabolism. Cancer Sci. 2009, 100, 821–827. [Google Scholar] [CrossRef] [PubMed]
- Metallo, C.M.; Gameiro, P.A.; Bell, E.L.; Mattaini, K.R.; Yang, J.; Hiller, K.; Jewell, C.M.; Johnson, Z.R.; Irvine, D.J.; Guarente, L.; et al. Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature 2011, 481, 380–384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirsch, H.A.; Iliopoulos, D.; Tsichlis, P.N.; Struhl, K. Metformin selectively targets cancer stem cells, and acts together with chemotherapy to block tumor growth and prolong remission. Cancer Res. 2009, 69, 7507–7511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.J.; Li, C.F.; Ruan, D.; He, J.; Montal, E.D.; Lorenz, S.; Girnun, G.D.; Chan, C.H. Non-proteolytic ubiquitination of Hexokinase 2 by HectH9 controls tumor metabolism and cancer stem cell expansion. Nat. Commun. 2019, 10, 2625. [Google Scholar] [CrossRef] [Green Version]
- Shibuya, K.; Okada, M.; Suzuki, S.; Seino, M.; Seino, S.; Takeda, H.; Kitanaka, C. Targeting the facilitative glucose transporter GLUT1 inhibits the self-renewal and tumor-initiating capacity of cancer stem cells. Oncotarget 2015, 6, 651–661. [Google Scholar] [CrossRef] [Green Version]
- Sato, A.; Sunayama, J.; Okada, M.; Watanabe, E.; Seino, S.; Shibuya, K.; Suzuki, K.; Narita, Y.; Shibui, S.; Kayama, T.; et al. Glioma-initiating cell elimination by metformin activation of FOXO3 via AMPK. Stem Cells Transl. Med. 2012, 1, 811–824. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kuramoto, K.; Yamamoto, M.; Suzuki, S.; Togashi, K.; Sanomachi, T.; Kitanaka, C.; Okada, M. Inhibition of the Lipid Droplet–Peroxisome Proliferator-Activated Receptor α Axis Suppresses Cancer Stem Cell Properties. Genes 2021, 12, 99. https://doi.org/10.3390/genes12010099
Kuramoto K, Yamamoto M, Suzuki S, Togashi K, Sanomachi T, Kitanaka C, Okada M. Inhibition of the Lipid Droplet–Peroxisome Proliferator-Activated Receptor α Axis Suppresses Cancer Stem Cell Properties. Genes. 2021; 12(1):99. https://doi.org/10.3390/genes12010099
Chicago/Turabian StyleKuramoto, Kenta, Masahiro Yamamoto, Shuhei Suzuki, Keita Togashi, Tomomi Sanomachi, Chifumi Kitanaka, and Masashi Okada. 2021. "Inhibition of the Lipid Droplet–Peroxisome Proliferator-Activated Receptor α Axis Suppresses Cancer Stem Cell Properties" Genes 12, no. 1: 99. https://doi.org/10.3390/genes12010099