Plasmodium vivax Cysteine-Rich Protective Antigen Polymorphism at Exon-1 Shows Recombination and Signatures of Balancing Selection




Abstract
:1. Introduction
2. Materials and Methods
2.1. Samples and Origin
2.2. DNA Extraction, PCR Amplification and Sequencing
2.3. Gene Sequences
2.4. Genetic Analysis of the Coding Gene
2.5. Linkage Disequilibrium (LD) Analysis and Mutations in the Intron
2.6. Exploration of Potential Residues Participating in B-cell Epitopes in CYRPA Amino Acid Sequences of P. vivax From Southern Mexico
3. Results
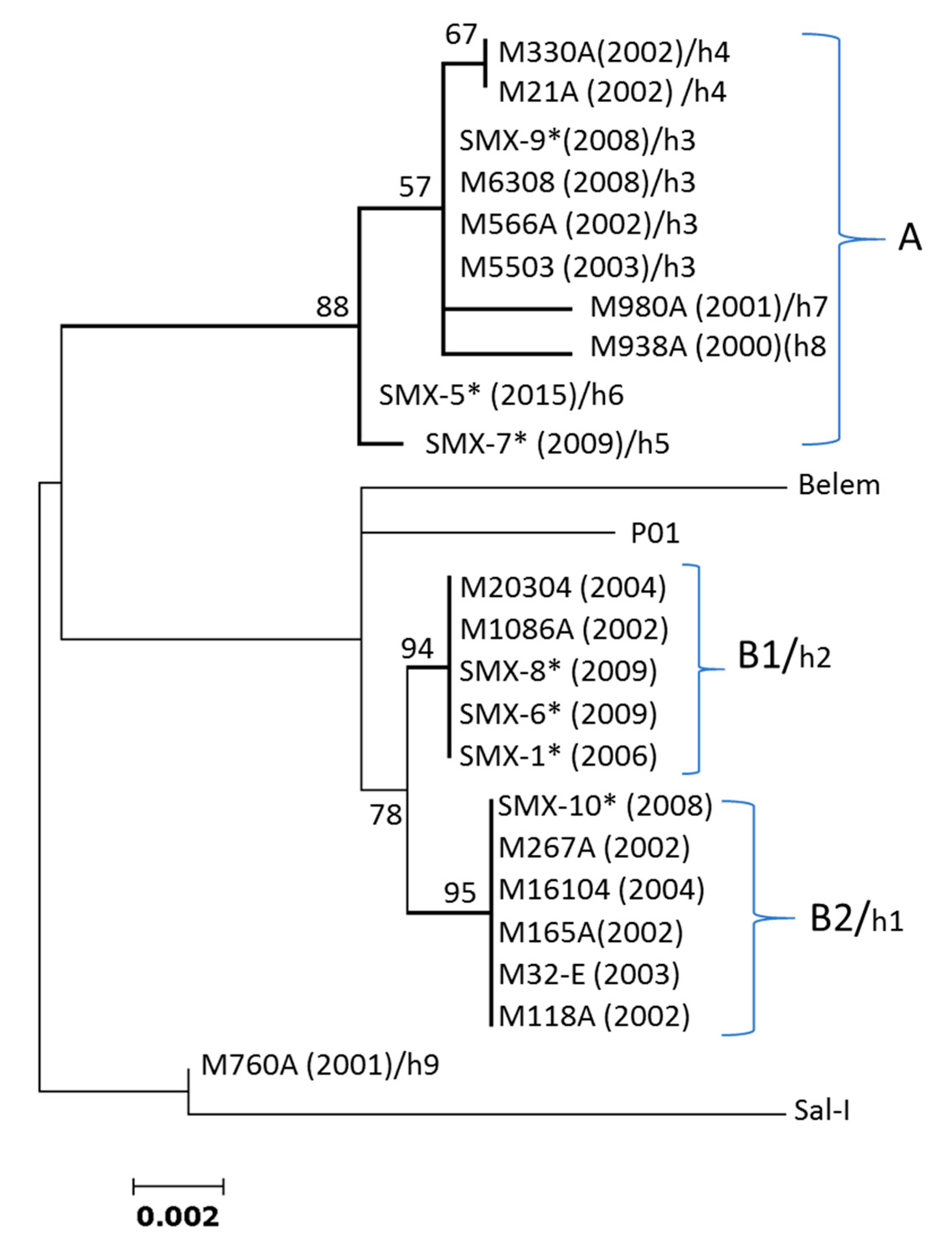
3.1. Pvcyrpa Polymorphism at the Coding Gene of Parasites from Southern Mexico
3.2. Sequence Diversity of the Pvcyrpa Coding Gene
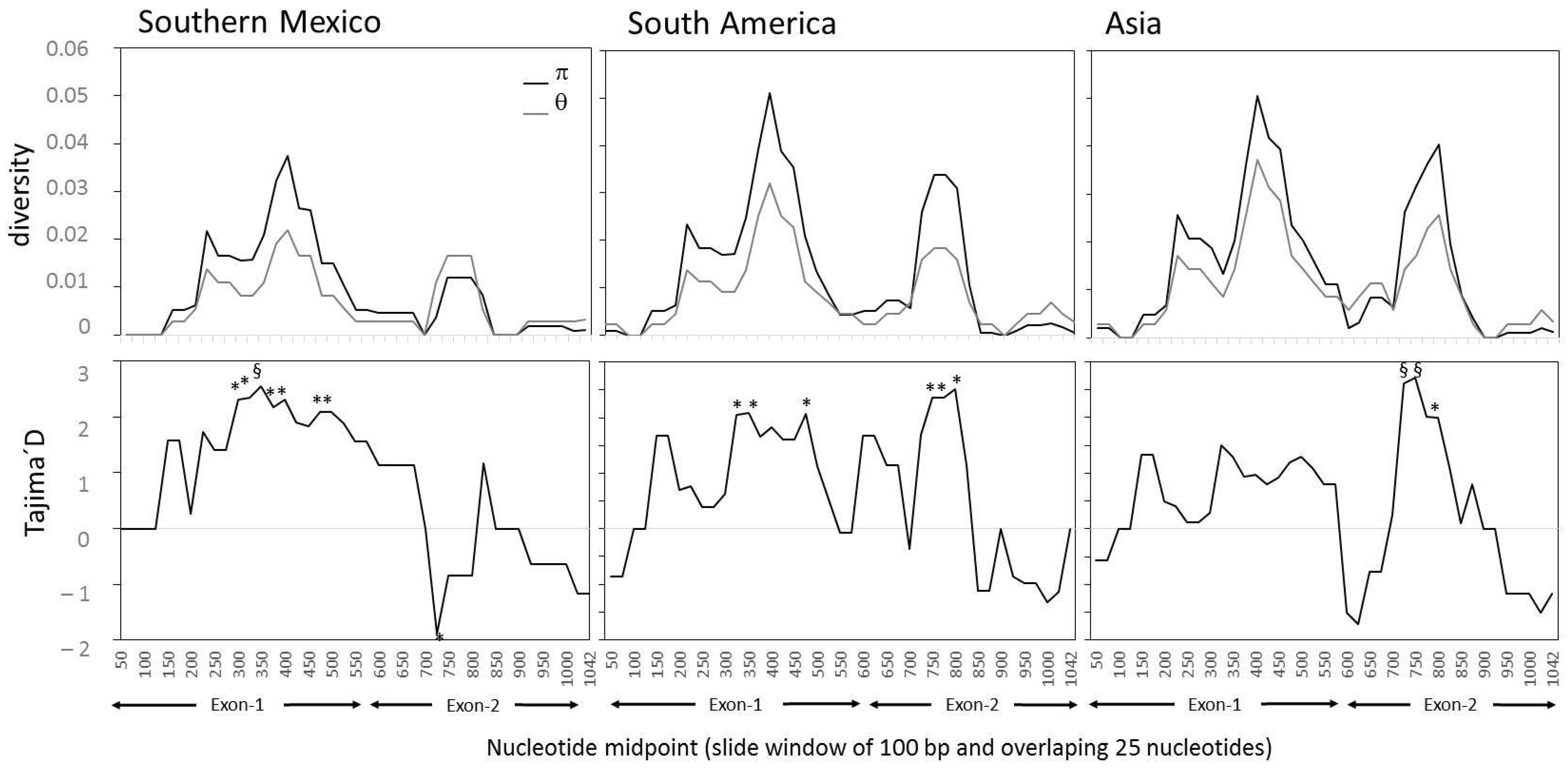
3.3. Neutrality Tests
3.4. Linkage Disequilibrium (LD) and Recombination
3.5. Prediction of Polymorphic Residues Potentially Participating in the B-Cell Epitope
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- WHO. Global Malaria Programme: WHO Global World Malaria Report 2019; WHO: Geneva, Switzerland, 2019; ISBN 9789241565721. [Google Scholar]
- Thera, M.A.; Plowe, C.V. Vaccines for malaria: How close are we? Annu. Rev. Med. 2012, 63, 345–357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- WHO. World Malaria Report 2017; WHO: Geneva, Switzerland, 2017; ISBN 9789241565523. [Google Scholar]
- França, C.T.; Hostetler, J.B.; Sharma, S.; White, M.T.; Lin, E.; Kiniboro, B.; Waltmann, A.; Darcy, A.W.; Suen, C.S.N.L.W.; Siba, P.; et al. An antibody screen of a plasmodium vivax antigen library identifies novel merozoite proteins associated with clinical protection. PLOS Negl. Trop. Dis. 2016, 10, e0004639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pires, C.V.; Alves, J.R.S.; Lima, B.A.S.; Paula, R.B.; Costa, H.L.; Torres, L.M.; Sousa, T.N.; Soares, I.S.; Sanchez, B.A.M.; Fontes, C.J.F.; et al. Blood-stage Plasmodium vivax antibody dynamics in a low transmission setting: A nine year follow-up study in the Amazon region. PLoS ONE 2018, 13, e0207244. [Google Scholar] [CrossRef] [PubMed]
- Volz, J.C.; Yap, A.; Sisquella, X.; Thompson, J.K.; Lim, N.T.; Whitehead, L.W.; Chen, L.; Lampe, M.; Tham, W.-H.; Wilson, D.; et al. Essential role of the PfRh5/PfRipr/CyRPA complex during plasmodium falciparum invasion of erythrocytes. Cell Host Microbe 2016, 20, 60–71. [Google Scholar] [CrossRef] [Green Version]
- Dreyer, A.M.; Matile, H.; Papastogiannidis, P.; Kamber, J.; Favuzza, P.; Voss, T.S.; Wittlin, S.; Pluschke, G. Passive immunoprotection of plasmodium falciparum-infected mice designates the CyRPA as candidate malaria vaccine antigen. J. Immunol. 2012, 188, 6225–6237. [Google Scholar] [CrossRef] [Green Version]
- Favuzza, P.; Blaser, S.; Dreyer, A.M.; Riccio, G.; Tamborrini, M.; Thoma, R.; Matile, H.; Pluschke, G. Generation of Plasmodium falciparum parasite-inhibitory antibodies by immunization with recombinantly-expressed CyRPA. Malar. J. 2016, 15, 161. [Google Scholar] [CrossRef] [Green Version]
- Tamborrini, M.; Hauser, J.; Schäfer, A.; Amacker, M.; Favuzza, P.; Kyungtak, K.; Fleury, S.; Pluschke, G. Vaccination with virosomally formulated recombinant CyRPA elicits protective antibodies against Plasmodium falciparum parasites in preclinical in vitro and in vivo models. NPJ Vaccines 2020, 5, 9. [Google Scholar] [CrossRef] [Green Version]
- Knuepfer, E.; Wright, K.E.; Prajapati, S.K.; Rawlinson, T.A.; Mohring, F.; Koch, M.; Lyth, O.R.; Howell, S.A.; Villasis, E.; Snijders, A.P.; et al. Divergent roles for the RH5 complex components, CyRPA and RIPR in human-infective malaria parasites. PLOS Pathog. 2019, 15, e1007809. [Google Scholar] [CrossRef]
- França, C.T.; White, M.T.; He, W.-Q.; Hostetler, J.; Brewster, J.; Frato, G.; Malhotra, I.; Gruszczyk, J.; Huon, C.; Lin, E.; et al. Identification of highly-protective combinations of Plasmodium vivax recombinant proteins for vaccine development. eLife 2017, 6. [Google Scholar] [CrossRef] [Green Version]
- WHO. World Malaria Report; World Health Organization: Geneva, Switzerland, 2015; Available online: http://www.who.int/malaria/publications/world-malaria-report-2015/report/en/ (accessed on 25 November 2020).
- González-Cerón, L.; Rodriguez, M.H.; Sandoval, M.A.; Santillán, F.; Galindo-Virgen, S.; Betanzos, A.F.; Rosales, A.F.; Palomeque, O.L. Effectiveness of combined chloroquine and primaquine treatment in 14 days versus intermittent single dose regimen, in an open, non-randomized, clinical trial, to eliminate Plasmodium vivax in southern Mexico. Malar. J. 2015, 14, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Mexican Official Standard for Epidemiological Surveillance, Prevention and Control of Vector-Borne Diseases. NOM-032-SSA2-2002. Health Secretary, Mexico; 2002. Available online: http://www.salud.gob.mx/unidades/cdi/nom/032ssa202.html (accessed on 20 November 2020).
- Rubio, J.M.; Benito, A.; Berzosa, P.J.; Roche, J.; Puente, S.; Subirats, M.; López-Vélez, R.; García, L.; Alvar, J. Usefulness of seminested multiplex PCR in surveillance of imported malaria in Spain. J. Clin. Microbiol. 1999, 37, 3260–3264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thompson, J.D.; Higgins, D.G.; Gibson, T.J. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994, 22, 4673–4680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hall, T.A. Bioedit: A user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucl. Acids Symp. Ser. 1999, 41, 95–98. [Google Scholar]
- Carlton, J.M.; Adams, J.H.; Silva, J.C.; Bidwell, S.L.; Lorenzi, H.; Caler, E.; Crabtree, J.; Angiuoli, S.V.; Merino, E.F.; Amedeo, P.; et al. Comparative genomics of the neglected human malaria parasite Plasmodium vivax. Nat. Cell Biol. 2008, 455, 757–763. [Google Scholar] [CrossRef] [PubMed]
- Flores-Alanis, A.; González-Cerón, L.; Santillán, F.; Ximenez, C.; Sandoval, M.A.; Cerritos, R. Temporal genetic changes in Plasmodium vivax apical membrane antigen 1 over 19 years of transmission in southern Mexico. Parasites Vectors 2017, 10, 1–12. [Google Scholar] [CrossRef]
- Hupalo, D.N.; Luo, Z.; Melnikov, A.; Sutton, P.L.; Rogov, P.; Escalante, A.; Vallejo, A.F.; Herrera, S.; Arévalo-Herrera, M.; Fan, Q.; et al. Population genomics studies identify signatures of global dispersal and drug resistance in Plasmodium vivax. Nat. Genet. 2016, 48, 953–958. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [Green Version]
- Rozas, J.; Ferrer-Mata, A.; Sánchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sánchez-Gracia, A. DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef]
- Fu, Y.X.; Li, W.H. Statistical tests of neutrality of mutations. Genetics 1993, 133, 693–709. [Google Scholar]
- Tajima, F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 1989, 123, 585–595. [Google Scholar]
- Murrell, B.; Moola, S.; Mabona, A.; Weighill, T.; Sheward, D.; Pond, S.L.K.; Scheffler, K. FUBAR: A fast, unconstrained bayesian approximation for inferring selection. Mol. Biol. Evol. 2013, 30, 1196–1205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nei, M.; Kumar, S. Molecular Evolution and Phylogenetics; Oxford University Press: New York, NY, USA, 2000. [Google Scholar]
- Tetteh, K.K.A.; Stewart, L.B.; Ochola-Oyier, L.I.; Amambua-Ngwa, A.; Thomas, A.W.; Marsh, K.; Weedall, G.D.; Conway, D.J. Prospective identification of malaria parasite genes under balancing selection. PLoS ONE 2009, 4, e5568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McDonald, J.H.; Kreitman, M. Adaptive protein evolution at the Adh locus in Drosophila. Nat. Cell Biol. 1991, 351, 652–654. [Google Scholar] [CrossRef] [PubMed]
- Tachibana, S.I.; Sullivan, S.A.; Kawai, S.; Nakamura, S.; Kim, H.R.; Goto, N.; Arisue, N.; Palacpac, N.M.Q.; Honma, H.; Yagi, M.; et al. Plasmodium cynomolgi genome sequences provide insight into Plasmodium vivax and the monkey malaria clade. Nat. Genet. 2012, 44, 1051–1055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chatterjee, S.; Mukhopadhyay, P.; Bandyopadhyay, R.; Dhal, P.; Biswal, D.; Bandyopadhyay, P.K. Molecular characterization and phylogenetic analysis of Plasmodium vivax, Plasmodium falciparum, Plasmodium ovale, Plasmodium malariae and Plasmodium cynomolgi. J. Parasit. Dis. 2016, 41, 230–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parker, J.M.R.; Guo, D.; Hodges, R.S. New hydrophilicity scale derived from high-performance liquid chromatography peptide retention data: Correlation of predicted surface residues with antigenicity and x-ray-derived accessible sites. Biochemistry 1986, 25, 5425–5432. [Google Scholar] [CrossRef]
- Jespersen, M.C.; Peters, B.; Nielsen, M.; Marcatili, P. BepiPred-2.0: Improving sequence-based B-cell epitope prediction using conformational epitopes. Nucleic Acids Res. 2017, 45, W24–W29. [Google Scholar] [CrossRef] [Green Version]
- Emini, E.A.; Hughes, J.V.; Perlow, D.S.; Boger, J. Induction of hepatitis A virus-neutralizing antibody by a virus-specific synthetic peptide. J. Virol. 1985, 55, 836–839. [Google Scholar] [CrossRef] [Green Version]
- González-Cerón, L.; Cerritos, R.; Corzo-Mancilla, J.; Santillán, F. Diversity and evolutionary genetics of the three major Plasmodium vivax merozoite genes participating in reticulocyte invasion in southern Mexico. Parasites Vectors 2015, 8, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Xu, Y.; Wong, W.; Thompson, J.K.; Healer, J.; Goddard-Borger, E.D.; Lawrence, M.C.; Cowman, A.F. Structural basis for inhibition of erythrocyte invasion by antibodies to Plasmodium falciparum protein CyRPA. eLife 2017, 6, 213. [Google Scholar] [CrossRef] [Green Version]
- González-Cerón, L.; Montoya, A.; Corzo-Gomez, J.; Cerritos, R.; Santillán, F.; Sandoval, M.A. Genetic diversity and natural selection of Plasmodium vivax multi-drug resistant gene (pvmdr1) in Mesoamerica. Malar. J. 2017, 16, 261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- González-Cerón, L.; Rodríguez, M.H.; Ovilla-Muñoz, M.T.; Santillán-Valenzuela, F.; Hernández-Ávila, J.E.; Rodríguez, M.C.; Barnetche, J.M.; Villarreal-Treviño, C. Ookinete-specific genes and 18S SSU rRNA evidenced in plasmodium vivax selection and adaptation by sympatric vectors. Front. Genet. 2020, 10, 1362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chitnis, C.E.; Sharma, A. Targeting the plasmodium vivax duffy-binding protein. Trends Parasitol. 2008, 24, 29–34. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez, S.; González-Cerón, L.; Montoya, A.; Sandoval, M.A.; Tórres, M.E.; Cerritos, R. Genetic structure of Plasmodium vivax in Nicaragua, a country in the control phase, based on the carboxyl terminal region of the merozoite surface protein-1. Infect. Genet. Evol. 2016, 40, 324–330. [Google Scholar] [CrossRef]
- Arnott, A.; Barry, A.E.; Reeder, J.C. Understanding the population genetics of Plasmodium vivax is essential for malaria control and elimination. Malar. J. 2012, 11, 14. [Google Scholar] [CrossRef] [Green Version]
- González-Cerón, L.; Mu, J.; Santillán, F.; Joy, D.A.; Sandoval, M.A.; Camas, G.; Su, X.Z.; Choy, E.V.; Torreblanca, R. Molecular and epidemiological characterization of Plasmodium vivax recurrent infections in southern Mexico. Parasites Vectors 2013, 6, 109. [Google Scholar] [CrossRef] [Green Version]
- Akey, J.M.; Eberle, M.A.; Rieder, M.J.; Carlson, C.S.; Shriver, M.D.; Nickerson, D.A.; Kruglyak, L. Population history and natural selection shape patterns of genetic variation in 132 genes. PLoS Biol. 2004, 2, e286. [Google Scholar] [CrossRef]
- Shen, H.-M.; Chen, S.-B.; Wang, Y.; Xu, B.; Abe, E.M.; Chen, J.-H. Genome-wide scans for the identification of Plasmodium vivax genes under positive selection. Malar. J. 2017, 16, 1–12. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer | Sequence (5′–3′) | Specificity | Reference |
|---|---|---|---|
| Reverse primer UNR | GACGGTATCTGATCGTCTTC | Universal | [15] |
| Forward primer PLF | AGTGTGTATCCAATCGAGTTTC | Plasmodium | [15] |
| Reverse primer VIR | AGGACTTCCAAGCCGAAGC | P. vivax | [15] |
| CYRPA_R1 | AGTTGGGATGTGCTACTGGAG | P. vivax | This article |
| CYRPA_F1 | TAAGTCTGCTTTCCTCTCTTGGG | P. vivax | This article |
| CYRPA_R2 | AGACTGGAAAGACGCAACGG | P. vivax | This article |
| CYRPA_F2 | TTGGAGGGACTTGTCCGGTT | P. vivax | This article |
| CYRPA_R3 | TGCTCTGTGTAGTAGAGG | P. vivax | This article |
| CYRPA_F3 | TTTTTCTCCCCTTGGGAGGCTAC | P. vivax | This article |
| CYRPA_R4 | GTGGAAAGAAGTGTGTGGAGGT | P. vivax | This article |
| CYRPA_F4 | TATGGGACTTTTGATGGTTG | P. vivax | This article |
| CYRPA_R5 | AACTGACTGGTATGAGTCC | P. vivax | This article |
| Haplotype | n | Exon-1 | Exon-2 | ||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Codon Number/Amino acid Residue | |||||||||||||||||||||||||
| 69 | 82 | 91 | 93 | 95 | 126 | 127 | 129 | 132 | 139 | 145 | 154 | 159 | 170 | 185 | 220 | 258 | 259 | 260 | 261 | 264 | 271 | 326 | 363 | ||
| Ala | Asn | Lys | Glu | Asn | Lys | Glu | Ile | Arg | Thr | Asn | Asp | Asp | Asp | Lys | Val | Leu1 | Glu | Glu | Pro | Ser | - | Asp | Thr | ||
| Nucleotides | |||||||||||||||||||||||||
| h1 | 5+1 | gcg | aac | Aag | gaa | Aat | aaa | gaa | atc | cgg | Acc | aac | gac | gac | Gac | aaa | gtc | cta | gaa | gag | ccg | agc | - | gat | acg |
| h2 | 2 + 3 | … | … | … | … | … | … | … | … | … | … | … | … | … | … | … | a.. | … | … | … | … | g.t | gaa | … | … |
| h3 | 3 + 1 | .g. | … | .c. | a.. | c.. | g.. | .g. | .g. | ..c | … | gg. | .c. | ..g | a.. | g.. | a.. | … | … | … | … | … | - | … | … |
| h4 | 2 + 0 | .g. | … | .c. | a.. | c.. | g.. | .g. | .g. | ..c | … | gg. | .c. | ..g | a.. | g.. | a.. | … | … | … | … | … | - | t.. | … |
| h5 | 0 + 1 | .g. | … | .c. | a.. | c.. | g.. | .g. | .g. | … | … | gg. | .c. | … | … | g.. | a.. | … | … | … | … | … | - | … | … |
| h6 | 0 + 1 | .g. | … | .c. | a.. | c.. | g.. | .g. | .g. | ..c | … | gg. | .c. | … | … | g.. | a.. | … | … | … | … | … | - | … | … |
| h7 | 1 + 0 | .g. | … | .c. | a.. | c.. | g.. | .g. | .g. | ..c | … | … | .c. | ..g | a.. | g.. | a.. | … | … | … | … | … | - | … | .g. |
| h8 | 1 + 0 | .g. | .g. | .c. | a.. | c.. | g.. | .g. | .g. | ..c | .a. | gg. | .c. | ..g | a.. | g.. | … | … | … | … | … | … | - | … | … |
| h9 | 1 + 0 | .g. | … | … | … | … | g.. | .g. | .g. | ..c | … | gg. | … | … | … | … | a.. | ..t | ..c | .t. | a.. | g.t | gaa | … | … |
| Amino acid change: | |||||||||||||||||||||||||
| Gly | Ser | Thr | Lys | His | Glu | Gly | Ser | * | Asn | Gly | Ala | Glu | Asn | Glu | Ile | * | Asp | Val | Thr | Gly | Glu | Tyr | Arg | ||
| Diversity | Entire Coding Gene: 1083 bp | exon-1 (605 bp) | exon-2 (478 bp) |
|---|---|---|---|
| Southern Mexico (n = 22) | |||
| SS | 25 | 16 | 9 |
| M | 25 | 16 | 9 |
| H | 9 | 7 | 6 |
| Hd ± SD | 0.861 ± 0.044 | 0.697 ± 0.0068 | 0.797 ± 0.046 |
| π ± DE | 0.0086 ± 0.0005 | 0.0124 ± 0.00086 | 0.004 ± 0.00084 |
| θw ± (DE nr, fr) | 0.0063± (0.0024, 0.0012) | 0.00727 (0.00294, 0.00182) | 0.00517 (0.0023, 0.00172) |
| Rm | 4 | 3 | 0 |
| South America (n = 45) | |||
| SS | 39 | 25 | 14 |
| M | 41 | 27 | 14 |
| H | 27 | 20 | 14 |
| Hd ± SD | 0.975 ± 0.009 | 0.92 ± 0.027 | 0.910 ± 0.018 |
| π ± DE | 0.01228 ± 0.00041 | 0.01467 ± 0.00055 | 0.00925 ± 0.00043 |
| θw ± (DE nr, fr) | 0.00824 (0.00263, 0.00132) | 0.00945 (0.00321, 0.00189) | 0.00670 (0.00254, 0.00179) |
| Rm | 11 | 8 | 2 |
| Asia (n = 19) | |||
| SS | 40 | 25 | 15 |
| M | 43 | 28 | 15 |
| H | 18 | 16 | 15 |
| Hd ± SD | 0.994 ± 0.019 | 0.982 ± 0.022 | 0.971 ± 0.027 |
| π ± DE | 0.0138 ± 0.00070 | 0.01627 ± 0.0010 | 0.01067 ± 0.00076 |
| θw ± (DE nr, fr) | 0.01057 (0.00392, 0.00167) | 0.0118 (0.000021; 0.0023) | 0.00898 (0.00375, 0.00232) |
| Rm | 12 | 8 | 3 |
| Parameters | Southern Mexico | South America | Asia | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Coding Gene | exon-1 | exon-2 | Coding Gene | exon-1 | exon-2 | Coding Gene | exon-1 | exon-2 | |
| Syn | 3 | 1 | 2 | 5 | 1 | 4 | 5 | 1 | 4 |
| Nonsyn | 22 | 15 | 7 | 32 | 22 | 10 | 31 | 20 3 | 11 |
| Tajima’s D (TjD) | 1.358 | 2.493 1 | −0.713 | 1.573 | 1.657 | 1.178 | 0.917 | 0.993 | 0.704 |
| TjD (syn) | 0.638 | 1.553 | −0.174 | 0.674 | 0.242 | 0.688 | 0.246 | −0.035 | 0.305 |
| TjD (nonsyn) | 1.401 | 2.425 1 | −0.805 | 1.652 | 1.697 | 1.210 | 1.003 | 1.040 | 0.787 |
| Fu & Li’D* | 0.042 | 0.870 | −1.284 | 1.3043 2 | 1.496 1 | 0.599 | 0.912 | 1.443 1 | −0.157 |
| Fu & Li’F* | 0.513 | 1.5765 2 | −1.297 | 1.6120 2 | 1.763 1 | 0.935 | 1.045 | 1.492 2 | 0.106 |
| Geographic Origin | Gene Fragment | Polymorphic Changes within P. vivax Groups | Z-Test (dN > dS) | P-Value | Fixed Differences between Species * | McDonald and Kreitman (Neutrality Index) | P-Value | ||
|---|---|---|---|---|---|---|---|---|---|
| Synonymous | Nonsynonymous | Synonymous | Nonsynonymous | ||||||
| Southern Mexico: | |||||||||
| Coding region | 2 | 21 | 2.399 | 0.009 | 82 | 84 | 10.25 | <0.001 | |
| n = 22 | exon-1 | 1 | 15 | 2.101 | 0.0189 | 48 | 56 | 12.857 | 0.0020 |
| exon-2 | 1 | 6 | 0.875 | 0.1912 | 33 | 27 | 7.333 | 0.0544 | |
| South America: | |||||||||
| Coding region | 4 | 32 | 2.8205 | 0.0028 | 81 | 82 | 7.902 | <0.001 | |
| n = 45 | exon-1 | 1 | 23 | 3.479 | <0.001 | 48 | 56 | 19.714 | <0.001 |
| exon-2 | 3 | 9 | 0.3909 | 0.3483 | 32 | 25 | 3.840 | 0.06238 | |
| Asia: | |||||||||
| Coding region | 4 | 30 | 2.883 | 0.0023 | 81 | 84 | 7.232 | <0.001 | |
| n = 19 | exon-1 | 1 | 20 | 3.529 | <0.001 | 48 | 56 | 17.143 | <0.001 |
| exon-2 | 3 | 10 | 0.803 | 0.2116 | 32 | 27 | 3.951 | 0.0647 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
González-Cerón, L.; Cebrián-Carmona, J.; Mesa-Valle, C.M.; García-Maroto, F.; Santillán-Valenzuela, F.; Garrido-Cardenas, J.A. Plasmodium vivax Cysteine-Rich Protective Antigen Polymorphism at Exon-1 Shows Recombination and Signatures of Balancing Selection. Genes 2021, 12, 29. https://doi.org/10.3390/genes12010029
González-Cerón L, Cebrián-Carmona J, Mesa-Valle CM, García-Maroto F, Santillán-Valenzuela F, Garrido-Cardenas JA. Plasmodium vivax Cysteine-Rich Protective Antigen Polymorphism at Exon-1 Shows Recombination and Signatures of Balancing Selection. Genes. 2021; 12(1):29. https://doi.org/10.3390/genes12010029
Chicago/Turabian StyleGonzález-Cerón, Lilia, José Cebrián-Carmona, Concepción M. Mesa-Valle, Federico García-Maroto, Frida Santillán-Valenzuela, and Jose Antonio Garrido-Cardenas. 2021. "Plasmodium vivax Cysteine-Rich Protective Antigen Polymorphism at Exon-1 Shows Recombination and Signatures of Balancing Selection" Genes 12, no. 1: 29. https://doi.org/10.3390/genes12010029