Building towards Precision Oncology for Pancreatic Cancer: Real-World Challenges and Opportunities


Abstract
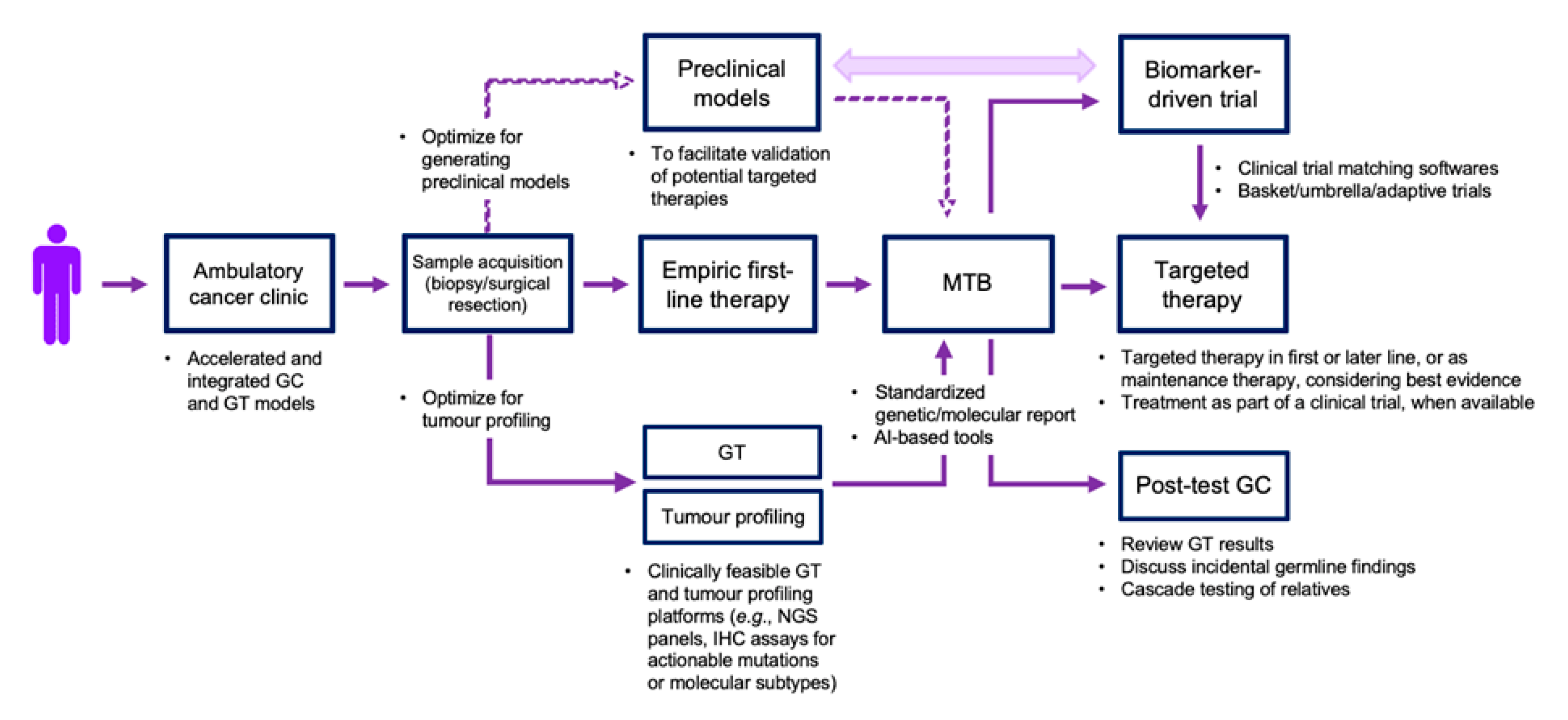
:1. Introduction
2. Inherited Predisposition to PDAC
2.1. Homologous Recombination Repair Deficient PDAC
2.2. Mismatch Repair Deficient PDAC
3. Somatic Alterations in PDAC: Drugging “Undruggable” Drivers
3.1. KRAS
3.2. Tumor Suppressor Genes
3.3. NTRK Gene Fusions
3.4. Transcriptomic Subtypes
3.5. Epigenetic Alterations in PDAC
4. Real-World Challenges in Implementing a PDAC Precision Oncology Program
4.1. Performing Germline Testing and Tumor Molecular Profiling
4.2. Returning and Interpreting Molecular Profiling Results
4.3. Validating and Accessing Biomarker-Driven Therapies
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2019. CA Cancer J Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, R.P.; Psarelli, E.-E.; Jackson, R.; Ghaneh, P.; Halloran, C.M.; Palmer, D.H.; Campbell, F.; Valle, J.W.; Faluyi, O.; O’Reilly, D.A.; et al. Patterns of recurrence after resection of pancreatic ductal adenocarcinoma. JAMA Surg. 2019, 154, 1038–1048. [Google Scholar] [CrossRef] [PubMed]
- Conroy, T.; Desseigne, F.; Ychou, M.; Bouché, O.; Guimbaud, R.; Bécouarn, Y. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N. Engl. J. Med. 2011, 364, 1817–1825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Von Hoff, D.D.; Ervin, T.; Arena, F.P.; Chiorean, E.G.; Infante, J.; Moore, M.; Seay, T.; Tjulandin, S.A.; Ma, W.W.; Saleh, M.N.; et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N. Engl. J. Med. 2013, 369, 1691–1703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burris, H.A.; Moore, M.J.; Andersen, J.; Green, M.R.; Rothenberg, M.L.; Modiano, M.R.; Cripps, M.C.; Portenoy, R.K.; Storniolo, A.M.; Tarassoff, P.; et al. Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: A randomized trial. J. Clin. Oncol. 1997, 15, 2403–2413. [Google Scholar] [CrossRef] [Green Version]
- Moore, M.; Goldstein, D.; Hamm, J.; Figer, A.; Hecht, J.R.; Gallinger, S.; Au, H.J.; Murawa, P.; Walde, D.; Wolff, R.A.; et al. Erlotinib plus gemcitabine compared with gemcitabine alone in patients with advanced pancreatic cancer: A Phase III trial of the national cancer institute of Canada clinical trials group. J. Clin. Oncol. 2007, 25, 1960–1966. [Google Scholar] [CrossRef]
- Wang-Gillam, A.; Li, C.P.; Bodoky, G.; Dean, A.; Shan, Y.S.; Jameson, G.; Macarulla, T.; Lee, K.-H.; Cunningham, D.; Blanc, J.F.; et al. Nanoliposomal irinotecan with fluorouracil and folinic acid in metastatic pancreatic cancer after previous gemcitabine-based therapy (NAPOLI-1): A global, randomised, open-label, phase 3 trial. Lancet 2016, 387, 545–557. [Google Scholar] [CrossRef]
- Neoptolemos, J.P.; Stocken, D.D.; Friess, H.; Bassi, C.; Dunn, J.A.; Hickey, H. A randomized trial of chemoradiotherapy and chemotherapy after resection of pancreatic cancer. N. Engl. J. Med. 2004, 350, 1200–1210. [Google Scholar] [CrossRef] [Green Version]
- Oettle, H.; Neuhaus, P.; Hochhaus, A.; Hartmann, J.T.; Gellert, K.; Ridwelski, K.; Niedergethmann, M.; Zülke, C.; Fahlke, J.; Arning, M.B.; et al. Adjuvant chemotherapy with gemcitabine and long-term outcomes among patients with resected pancreatic cancer: The CONKO-001 randomized trial. JAMA 2013, 310, 1473–1481. [Google Scholar] [CrossRef] [Green Version]
- Neoptolemos, J.P.; Palmer, D.H.; Ghaneh, P.; Psarelli, E.E.; Valle, J.W.; Halloran, C.M. Comparison of adjuvant gemcitabine and capecitabine with gemcitabine monotherapy in patients with resected pancreatic cancer (ESPAC-4): A multicentre, open-label, randomised, phase 3 trial. Lancet 2017, 389, 1011–1024. [Google Scholar] [CrossRef]
- Conroy, T.; Hammel, P.; Hebbar, M.; Ben Abdelghani, M.; Wei, A.C.; Raoul, J.L.; Choné, L.; Francois, E.; Artru, P.; Biagi, J.J.; et al. FOLFIRINOX or gemcitabine as adjuvant therapy for pancreatic cancer. N. Engl. J. Med. 2018, 379, 2395–2406. [Google Scholar] [CrossRef] [PubMed]
- Petersen, G.M. Familial pancreatic cancer. Semin. Oncol. 2016, 43, 548–553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klein, A.P. Prospective risk of pancreatic cancer in familial pancreatic cancer kindreds. Cancer Res. 2004, 64, 2634–2638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lowery, M.A.; Wong, W.; Jordan, E.J.; Lee, J.W.; Kemel, Y.; Vijai, J.; Mandelker, D.; Zehir, A.; Capanu, M.; Salo-Mullen, E.; et al. Prospective evaluation of germline alterations in patients with exocrine pancreatic neoplasms. J. Natl. Cancer Inst. 2018, 110, 1067–1074. [Google Scholar] [CrossRef] [Green Version]
- Holter, S.; Borgida, A.; Dodd, A.; Grant, R.; Semotiuk, K.; Hedley, D.; Dhani, N.; Narod, S.; Akbari, M.; Moore, M.; et al. Germline BRCA mutations in a large clinic-based cohort of patients with pancreatic adenocarcinoma. J. Clin. Oncol. 2015, 33, 3124–3129. [Google Scholar] [CrossRef]
- Grant, R.C.; Selander, I.; Connor, A.A.; Selvarajah, S.; Borgida, A.; Briollais, L. Prevalence of germline mutations in cancer predisposition genes in patients with pancreatic cancer. Gastroenterology 2015, 148, 556–564. [Google Scholar] [CrossRef] [Green Version]
- Hu, C.; Hart, S.N.; Polley, E.C.; Gnanaolivu, R.; Shimelis, H.; Lee, K.Y.; Lilyquist, J.; Na, J.; Moore, R.M.; Antwi, S.O.; et al. Association between inherited germline mutations in cancer predisposition genes and risk of pancreatic cancer. JAMA 2018, 319, 2401–2409. [Google Scholar] [CrossRef]
- Ferrone, C.R.; Levine, D.A.; Tang, L.H.; Allen, P.J.; Jarnagin, W.; Brennan, M.F.; Offit, K.; Robson, M.E. BRCA germline mutations in jewish patients with pancreatic adenocarcinoma. J. Clin. Oncol. 2009, 27, 433–438. [Google Scholar] [CrossRef] [Green Version]
- Smith, A.L.; Wong, C.; Cuggia, A.; Borgida, A.; Holter, S.; Hall, A.; Connor, A.A.; Bascuñana, C.; Asselah, J.; Bouganim, N.; et al. Reflex testing for germline BRCA1, BRCA2, PALB2, and ATM mutations in pancreatic cancer: Mutation prevalence and clinical outcomes from two canadian research registries. JCO Precis. Oncol. 2018, 2, 1–16. [Google Scholar] [CrossRef]
- Nanda, N.; Roberts, N.J. ATM serine/threonine kinase and its role in pancreatic risk. Genes 2020, 11, 108. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Heyer, W.D. Homologous recombination in DNA repair and DNA damage tolerance. Cell Res. 2008, 18, 99–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lord, C.J.; Ashworth, A. The DNA damage response and cancer therapy. Nature 2012, 481, 287–294. [Google Scholar] [CrossRef] [PubMed]
- Helleday, T. The underlying mechanism for the PARP and BRCA synthetic lethality: Clearing up the misunderstandings. Mol. Oncol. 2011, 5, 387–393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pommier, Y.G.; O’Connor, M.J.; De Bono, J. Laying a trap to kill cancer cells: PARP inhibitors and their mechanisms of action. Sci. Transl. Med. 2016, 8, 362ps17. [Google Scholar] [CrossRef]
- Andrei, A.Z.; Hall, A.; Smith, A.L.; Bascuñana, C.; Malina, A.; Connor, A. Increased in vitro and in vivo sensitivity of BRCA2-associated pancreatic cancer to the poly(ADP-ribose) polymerase-1/2 inhibitor BMN 673. Cancer Lett. 2015, 364, 8–16. [Google Scholar] [CrossRef]
- Wang, Y.; Park, J.Y.P.; Pacis, A.; Denroche, R.E.; Jang, G.H.; Zhang, A.; Cuggia, A.; Domecq, C.; Monlong, J.; Raitses-Gurevich, M.; et al. A preclinical trial and molecularly-annotated patient cohort identify predictive biomarkers in homologous recombination deficient pancreatic cancer. Clin. Cancer Res. 2020. [Google Scholar] [CrossRef]
- Golan, T.; Stossel, C.; Atias, D.; Buzhor, E.; Halperin, S.; Cohen, K.; Raitses-Gurevich, M.; Glick, Y.; Raskin, S.; Yehuda, D.; et al. Recapitulating the clinical scenario of BRCA-associated pancreatic cancer in pre-clinical models. Int. J. Cancer 2018, 143, 179–183. [Google Scholar] [CrossRef] [Green Version]
- Golan, T.; Kanji, Z.S.; Epelbaum, R.; Devaud, N.; Dagan, E.; Holter, S.; Aderka, D.; Paluch-Shimon, S.; Kaufman, B.; Gershoni-Baruch, R.; et al. Overall survival and clinical characteristics of pancreatic cancer in BRCA mutation carriers. Br. J. Cancer 2014, 111, 1132–1138. [Google Scholar] [CrossRef]
- Golan, T.; Hammel, P.; Reni, M.; Van Cutsem, E.; Macarulla, T.; Hall, M.J.; Park, J.O.; Hochhauser, D.; Arnold, D.; Oh, Y.; et al. Maintenance olaparib for germline BRCA-mutated metastatic pancreatic cancer. N. Engl. J. Med. 2019, 381, 317–327. [Google Scholar] [CrossRef]
- O’Reilly, E.M.; Lee, J.W.; Zalupski, M.; Capanu, M.; Park, J.; Golan, T.; Tahover, E.; Lowery, M.A.; Chou, J.F.; Sahai, V.; et al. Randomized, multicenter, phase II trial of gemcitabine and cisplatin with or without veliparib in patients with pancreas adenocarcinoma and a germline BRCA/PALB2 mutation. J. Clin. Oncol. 2020, 38, 1378–1388. [Google Scholar] [CrossRef]
- Knudson, A.G. Mutation and cancer: Statistical study of retinoblastoma. Proc. Natl. Acad. Sci. USA 1971, 68, 820–823. [Google Scholar] [CrossRef] [Green Version]
- Polak, P.; Kim, J.; Braunstein, L.Z.; Tiao, G.; Karlic, R.; Rosebrock, D.; Livitz, D.; Kübler, K.; Mouw, K.W.; Haradhavala, N.J.; et al. A mutational signature reveals alterations underlying deficient homologous recombination repair in breast cancer. Nat. Genet. 2017, 49, 1476–1486. [Google Scholar] [CrossRef] [PubMed]
- Davies, H.; Glodzik, D.; Morganella, S.; Yates, L.R.; Staaf, J.; Zou, X.; Ramakrishna, M.; Martin, S.; Boyault, S.; Sieuwerts, A.; et al. HRDetect is a predictor of BRCA1 and BRCA2 deficiency based on mutational signatures. Nat. Med. 2017, 23, 517–525. [Google Scholar] [CrossRef] [PubMed]
- Park, W.; Chen, J.; Chou, J.F.; Varghese, A.M.; Yu, K.H.; Wong, W.; Capanu, M.; Balachandran, V.; McIntyre, C.A.; El Dika, I.; et al. Genomic methods identify homologous recombination deficiency in pancreas adenocarcinoma and optimize treatment selection. Clin. Cancer Res. 2020. [Google Scholar] [CrossRef]
- Hughley, R.; Karlic, R.; Joshi, H.; Turnbull, C.; Foulkes, W.D.; Polak, P. Etiologic index—A case-only measure of BRCA1/2-associated cancer risk. N. Engl. J. Med. 2020, 383, 286–288. [Google Scholar] [CrossRef]
- Mersch, J.; Jackson, M.A.; Park, M.; Nebgen, D.R.; Peterson, S.K.; Singletary, C.N.; Arun, B.K.; Litton, J.K. Cancers associated with BRCA1 and BRCA2 mutations other than breast and ovarian. Cancer 2015, 121, 269–275. [Google Scholar] [CrossRef] [Green Version]
- Kastrinos, F.; Mukherjee, B.; Tayob, N.; Wang, F.; Sparr, J.; Raymond, V.M. Risk of pancreatic cancer in families with Lynch syndrome. JAMA 2009, 302, 1790–1795. [Google Scholar] [CrossRef]
- Blando, J.; Sharma, A.; Higa, M.G.; Zhao, H.; Vence, L.; Yadav, S.S.; Kim, J.; Sepulveda, A.M.; Sharp, M.; Maitra, A.; et al. Comparison of immune infiltrates in melanoma and pancreatic cancer highlights VISTA as a potential target in pancreatic cancer. Proc. Natl. Acad. Sci. USA 2019, 116, 1692–1697. [Google Scholar] [CrossRef] [Green Version]
- Connor, A.A.; Denroche, R.E.; Jang, G.H.; Timms, L.; Kalimuthu, S.N.; Selander, I.; McPherson, T.; Wilson, G.W.; Chan-Seng-Yue, M.A.; Borozan, I.; et al. Association of distinct mutational signatures with correlates of increased immune activity in pancreatic ductal adenocarcinoma. JAMA Oncol. 2017, 3, 774–783. [Google Scholar] [CrossRef]
- Riaz, N.; Morris, L.; Havel, J.J.; Makarov, V.; Desrichard, A.; Chan, T. The role of neoantigens in response to immune checkpoint blockade. Int. Immunol. 2016, 28, 411–419. [Google Scholar] [CrossRef] [Green Version]
- Le, D.T.; Durham, J.N.; Smith, K.N.; Wang, H.; Bartlett, B.R.; Aulakh, L.K. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017, 357, 409–413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, Z.I.; Shia, J.; Stadler, Z.K.; Varghese, A.M.; Capanu, M.; Salo-Mullen, E.; Lowery, M.A.; Diaz, L.A.; Mandelker, D.; Yu, K.H.; et al. Evaluating mismatch repair deficiency in pancreatic adenocarcinoma: Challenges and recommendations. Clin. Cancer Res. 2018, 24, 1326–1336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marabelle, A.; Le, D.T.; Ascierto, P.A.; Di Giacomo, A.M.; De Jesus-Acosta, A.; Delord, J.P.; Geva, R.; Gottfried, M.; Penel, N.; Hansen, A.R.; et al. Efficacy of pembrolizumab in patients with noncolorectal high microsatellite instability/mismatch repair-deficient cancer: Results from the phase II KEYNOTE-158 study. J. Clin. Oncol. 2019, 38, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Singhi, A.D.; George, B.; Greenbowe, J.R.; Chung, J.; Suh, J.; Maitra, A.; Klempner, S.J.; Hendifar, A.; Milind, J.M.; Golan, T.; et al. Real-Time Targeted Genome Profile Analysis of Pancreatic Ductal Adenocarcinomas Identifies Genetic Alterations That Might Be Targeted With Existing Drugs or Used as Biomarkers. Gastroenterology 2019, 156, 2242–2253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCormick, F. KRAS as a Therapeutic Target. Clin. Cancer Res. 2015, 21, 1797–1801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yaeger, R.; Corcoran, R.B. Targeting alterations in the RAF–MEK pathway. Cancer Discov. 2019, 9, 329–341. [Google Scholar] [CrossRef] [Green Version]
- Canon, J.; Rex, K.; Saiki, A.Y.; Mohr, C.; Cooke, K.; Bagal, D.; Gaida, K.; Holt, T.; Knutson, C.G.; Koppada, N.; et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature 2019, 575, 217–223. [Google Scholar] [CrossRef]
- Govindan, R.; Fakih, M.; Price, T.; Falchook, G.; Desai, J.; Kuo, J. OA02.02 Phase 1 study of safety, tolerability, PK and efficacy of AMG 510, a novel KRASG12C inhibitor, evaluated in NSCLC. J. Thorac. Oncol. 2019, 14, S208. [Google Scholar] [CrossRef]
- Hong, D.S.; Kuo, J.; Sacher, A.G.; Barlesi, F.; Besse, B.; Kuboki, Y.; Dy, G.K.; Dembla, V.; Krauss, J.C.; Burns, T.F.; et al. CodeBreak 100: Phase I study of AMG 510, a novel KRASG12C inhibitor, in patients (pts) with advanced solid tumors other than non-small cell lung cancer (NSCLC) and colorectal cancer (CRC). J. Clin. Oncol. 2020, 38, 3511. [Google Scholar] [CrossRef]
- Hofmann, M.H.; Gmachl, M.; Ramharter, J.; Savarese, F.; Gerlach, D.; Marszalek, J.R.; Sanderson, M.P.; Kessler, D.; Trapani, F.; Arnhof, H.; et al. BI-3406, a potent and selective SOS1::KRAS interaction inhibitor, is effective in KRAS-driven cancers through combined MEK inhibition. Cancer Discov. 2020. [Google Scholar] [CrossRef]
- Guan, M.; Bender, R.J.; Pishvaian, M.J.; Halverson, D.C.; Tuli, R.; Klempner, S.J.; Wainberg, Z.A.; Singhi, A.D.; Petricoin, E.; Hendifar, A.E. Molecular and clinical characterization of BRAF mutations in pancreatic ductal adenocarcinomas (PDACs). J. Clin. Oncol. 2018, 36, 214. [Google Scholar] [CrossRef]
- Chou, A.; Waddell, N.; Cowley, M.J.; Gill, A.J.; Chang, D.K.; Patch, A.-M.; Nones, K.; Wu, J.; Pinese, M.; Johns, A.L.; et al. Clinical and molecular characterization of HER2 amplified-pancreatic cancer. Genome Med. 2013, 5, 78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kvederaviciute, K.; Meskinyte, I.; Meskinyte-Kausiliene, E.; Skeberdyte, A.; Cicenas, J. KRAS, TP53, CDKN2A, SMAD4, BRCA1, and BRCA2 mutations in pancreatic cancer. Cancers 2017, 9, 42. [Google Scholar]
- Chen, J. The cell-cycle arrest and apoptotic functions of p53 in tumor initiation and progression. Cold Spring Harb. Perspect. Med. 2016, 6, a026104. [Google Scholar] [CrossRef]
- Cluzeau, T.; Sebert, M.; Rahmé, R.; Cuzzubbo, S.; Walter-Petrich, A.; Che, J.L.; Peterlin, P.; Beve, B.; Attalah, H.; Chermat, F.; et al. APR-246 combined with azacitidine (AZA) in TP53 mutated myelodysplastic syndrome (MDS) and acute myeloid leukemia (AML). A phase 2 study by the groupe francophone des myélodysplasies (GFM). Blood 2019, 134, 677. [Google Scholar] [CrossRef]
- Waddell, N.; Initiative, A.P.C.G.; Pajic, M.; Patch, A.M.; Chang, D.K.; Kassahn, K.S.; Bailey, P.; Johns, A.L.; Miller, D.K.; Nones, K.; et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature 2015, 518, 495–501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turner, N.C.; Slamon, D.J.; Ro, J.; Bondarenko, I.; Im, S.-A.; Masuda, N.; Colleoni, M.; DeMichele, A.; Loi, S.M.; Verma, S.; et al. Overall survival with palbociclib and fulvestrant in advanced breast cancer. N. Engl. J. Med. 2018, 379, 1926–1936. [Google Scholar] [CrossRef]
- Schettini, F.; De Santo, I.; Rea, C.G.; De Placido, P.; Formisano, L.; Giuliano, M. CDK 4/6 inhibitors as single agent in advanced solid tumors. Front Oncol. 2018, 8, 608. [Google Scholar] [CrossRef] [Green Version]
- Salvador-Barbero, B.; Álvarez-Fernández, M.; Zapatero-Solana, E.; Bakkali El, A.; Menéndez, M.D.C.; López-Casas, P.P. CDK4/6 inhibitors impair recovery from cytotoxic chemotherapy in pancreatic adenocarcinoma. Cancer Cell 2020, 37, 340–346. [Google Scholar] [CrossRef]
- Yingling, J.M.; McMillen, W.T.; Yan, L.; Huang, H.; Sawyer, J.S.; Graff, J.; Clawson, D.K.; Britt, K.S.; Anderson, B.D.; Beight, D.W.; et al. Preclinical assessment of galunisertib (LY2157299 monohydrate), a first-in-class transforming growth factor-β receptor type I inhibitor. Oncotarget 2017, 9, 6659–6677. [Google Scholar] [CrossRef] [Green Version]
- Holmgaard, R.B.; Schaer, D.A.; Li, Y.; Castaneda, S.P.; Murphy, M.Y.; Xu, X.; Inigo, I.; Dobkin, J.; Manro, J.R.; Iversen, P.W.; et al. Targeting the TGFβ pathway with galunisertib, a TGFβRI small molecule inhibitor, promotes anti-tumor immunity leading to durable, complete responses, as monotherapy and in combination with checkpoint blockade. J. Immunother. Cancer 2018, 6, 47. [Google Scholar] [CrossRef] [PubMed]
- Melisi, D.; Garcia-Carbonero, R.; Macarulla, T.; Pezet, D.; Deplanque, G.; Fuchs, M.; Trojan, J.; Oettle, H.; Kozloff, M.; Cleverly, A.; et al. Galunisertib plus gemcitabine vs. gemcitabine for first-line treatment of patients with unresectable pancreatic cancer. Br. J. Cancer 2018, 119, 1208–1214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsieh, Y.Y.; Liu, T.P.; Chou, C.J.; Chen, H.Y.; Lee, K.H.; Yang, P.M. Integration of bioinformatics resources reveals the therapeutic benefits of gemcitabine and cell cycle intervention in SMAD4-deleted pancreatic ductal adenocarcinoma. Genes 2019, 10, 766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solomon, J.; Benayed, R.; Hechtman, J.; Ladanyi, M. Identifying patients with NTRK fusion cancer. Ann. Oncol. 2019, 30, viii16–viii22. [Google Scholar] [CrossRef] [Green Version]
- Pishvaian, M.J.; Rolfo, C.D.; Liu, S.V.; Multani, P.S.; Maneval, E.C.; Garrido-Laguna, I. Clinical benefit of entrectinib for patients with metastatic pancreatic cancer who harbor NTRK and ROS1 fusions. J. Clin. Oncol. 2018, 36, 521. [Google Scholar] [CrossRef]
- Patel, M.; Siena, S.; Demetri, G.; Doebele, R.; Chae, Y.; Conkling, P. O-3 Efficacy and safety of entrectinib in NTRK fusion-positive gastrointestinal cancers: Updated integrated analysis of three clinical trials (STARTRK-2, STARTRK-1 and ALKA-372-001). Ann. Oncol. 2020, 31, 232–233. [Google Scholar] [CrossRef]
- Demols, A.; Perez-Casanova, L.; Rocq, L.; Charry, M.; De Nève, N.; Verrellen, A. NTRK gene fusions in bilio-pancreatic cancers. J. Clin. Oncol. 2020, 38 (Suppl. 15), e16664. [Google Scholar] [CrossRef]
- Collisson, E.A.; Sadanandam, A.; Olson, P.; Gibb, W.J.; Truitt, M.; Gu, S.; Cooc, J.; Weinkle, J.; Kim, G.E.; Jakkula, L.; et al. Subtypes of pancreatic ductal adenocarcinoma and their differing responses to therapy. Nat. Med. 2011, 17, 500–503. [Google Scholar] [CrossRef]
- Moffitt, R.A.; Marayati, R.; Flate, E.L.; Volmar, K.E.; Loeza, S.G.H.; Hoadley, K.A.; Rashid, N.U.; Williams, L.A.; Eaton, S.C.; Chung, A.H.; et al. Virtual microdissection identifies distinct tumor- and stroma-specific subtypes of pancreatic ductal adenocarcinoma. Nat. Genet. 2015, 47, 1168–1178. [Google Scholar] [CrossRef]
- Bailey, P.; Initiative, A.P.C.G.; Chang, D.K.; Nones, K.; Johns, A.L.; Patch, A.-M.; Gingras, M.-C.; Miller, D.K.; Christophi, C.; Bruxner, T.J.; et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 2016, 531, 47–52. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research Network; Raphael, B.J.; Hruban, R.H.; Aguirre, A.J.; Moffitt, R.A.; Yeh, J.J.; Stewart, C.; Robertson, A.G.; Cherniack, A.D.; Gupta, M. Integrated genomic characterization of pancreatic ductal adenocarcinoma. Cancer Cell 2017, 32, 185–203.e13. [Google Scholar] [CrossRef] [PubMed]
- Aung, K.L.; Fischer, S.E.; Denroche, R.E.; Jang, G.H.; Dodd, A.; Creighton, S.; Southwood, B.; Liang, S.B.; Chadwick, D.; Zhang, A.; et al. Genomics-driven precision medicine for advanced pancreatic cancer: Early results from the COMPASS trial. Clin. Cancer Res. 2017, 24, 1344–1354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinelli, P.; Pau, E.C.D.S.; Cox, T.; Sainz, B.; Dusetti, N.; Greenhalf, W.; Rinaldi, L.; Costello, E.; Ghaneh, P.; Malats, N.; et al. GATA6 regulates EMT and tumour dissemination, and is a marker of response to adjuvant chemotherapy in pancreatic cancer. Gut 2016, 66, 1665–1676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Kane, G.M.; Grünwald, B.T.; Jang, G.H.; Masoomian, M.; Picardo, S.; Grant, R.C.; Denroche, R.E.; Zhang, A.; Wang, Y.; Lam, B.; et al. GATA6 expression distinguishes classical and basal-like subtypes in advanced pancreatic cancer. Clin. Cancer Res. 2020. [Google Scholar] [CrossRef] [Green Version]
- Chan-Seng-Yue, M.; Kim, J.C.; Wilson, G.W.; Ng, K.; Figueroa, E.F.; O’Kane, G.M. Transcription phenotypes of pancreatic cancer are driven by genomic events during tumor evolution. Nat. Genet. 2020, 52, 231–240. [Google Scholar] [CrossRef]
- Hong, S.M.; Kelly, D.; Griffith, M.; Omura, N.; Li, A.; Li, C.P.; Hruban, R.H.; Goggins, M. Multiple genes are hypermethylated in intraductal papillary mucinous neoplasms of the pancreas. Mod. Pathol. 2008, 21, 1499–1507. [Google Scholar] [CrossRef]
- Li, A.; Omura, N.; Hong, S.M.; Goggins, M. Pancreatic cancer DNMT1 expression and sensitivity to DNMT1 inhibitors. Cancer Biol. Ther. 2010, 9, 321–329. [Google Scholar] [CrossRef] [Green Version]
- Lomberk, G.L.; Blum, Y.; Nicolle, R.; Nair, A.; Gaonkar, K.S.; Marisa, L.; Mathison, A.; Sun, Z.F.; Yan, H.; Elarouci, N.; et al. Distinct epigenetic landscapes underlie the pathobiology of pancreatic cancer subtypes. Nat. Commun. 2018, 9, 1–10. [Google Scholar] [CrossRef]
- Gailhouste, L.; Liew, L.C.; Hatada, I.; Nakagama, H.; Ochiya, T. Epigenetic reprogramming using 5-azacytidine promotes an anti-cancer response in pancreatic adenocarcinoma cells. Cell Death Dis. 2018, 9, 412–468. [Google Scholar] [CrossRef] [Green Version]
- Zagorac, S.; Alcala, S.; Bayón, G.F.; Kheir, T.B.; Schoenhals, M.; González-Neira, A.; Fraga, M.F.; Aicher, A.; Heeschen, C.; Sainz, B. DNMT1 inhibition reprograms pancreatic cancer stem cells via upregulation of the miR-17-92 cluster. Cancer Res. 2016, 76, 4546–4558. [Google Scholar] [CrossRef] [Green Version]
- Nicolle, R.; Blum, Y.; Marisa, L.; Loncle, C.; Gayet, O.; Moutardier, V.; Turrini, O.; Giovannini, M.; Bian, B.; Bigonnet, M.; et al. Pancreatic adenocarcinoma therapeutic targets revealed by tumor-stroma cross-talk analyses in patient-derived xenografts. Cell Rep. 2017, 21, 2458–2470. [Google Scholar] [CrossRef] [PubMed]
- Sato, N.; Fukushima, N.; Hruban, R.H.; Goggins, M. CpG island methylation profile of pancreatic intraepithelial neoplasia. Mod. Pathol. 2007, 21, 238–244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Natale, F.; Vivo, M.; Falco, G.; Angrisano, T. Deciphering DNA methylation signatures of pancreatic cancer and pancreatitis. Clin. Epigenet. 2019, 11, 132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eissa, M.A.L.; Lerner, L.; Abdelfatah, E.; Shankar, N.; Canner, J.K.; Hasan, N.M.; Yaghoobi, V.; Huang, B.; Kerner, Z.; Takaesu, F.; et al. Promoter methylation of ADAMTS1 and BNC1 as potential biomarkers for early detection of pancreatic cancer in blood. Clin. Epigene. 2019, 11, 59. [Google Scholar] [CrossRef]
- Brancaccio, M.; Natale, F.; Falco, G.; Angrisano, T. Cell-free DNA methylation: The new frontiers of pancreatic cancer biomarkers’ discovery. Genes 2019, 11, 14. [Google Scholar] [CrossRef] [Green Version]
- Cheng, D.T.; Mitchell, T.N.; Zehir, A.; Shah, R.; Benayed, R.; Syed, A.; Chandramohan, R.; Liu, Z.Y.; Won, H.H.; Scott, S.N.; et al. Memorial sloan kettering-integrated mutation profiling of actionable cancer targets (MSK-IMPACT): A hybridization capture-based next-generation sequencing clinical assay for solid tumor molecular oncology. J. Mol. Diagn. 2015, 17, 251–264. [Google Scholar] [CrossRef]
- Allegretti, M.; Fabi, A.; Buglioni, S.; Martayan, A.; Conti, L.; Pescarmona, E.; Ciliberto, G.; Giacomini, P. Tearing down the walls: FDA approves next generation sequencing (NGS) assays for actionable cancer genomic aberrations. J. Exp. Clin. Cancer Res. 2018, 37, 1–3. [Google Scholar] [CrossRef] [Green Version]
- Tempero, M.A. NCCN guidelines updates: Pancreatic cancer. J. Natl. Compr. Cancer Netw. 2019, 17, 603–605. [Google Scholar]
- Mosele, F.; Remon, J.; Mateo, J.; Westphalen, C.; Barlesi, F.; Lolkema, M.; Normanno, N.; Scarpa, A.; Robson, M.; Meric-Bernstam, F.; et al. Recommendations for the use of next-generation sequencing (NGS) for patients with metastatic cancers: A report from the ESMO precision medicine working group. Ann. Oncol. 2020. [Google Scholar] [CrossRef]
- Shabihkhani, M.; Lucey, G.M.; Wei, B.; Mareninov, S.; Lou, J.J.; Vinters, H.V.; Singer, E.J.; Cloughesy, T.F.; Yong, W.H. The procurement, storage, and quality assurance of frozen blood and tissue biospecimens in pathology, biorepository, and biobank settings. Clin. Biochem. 2014, 47, 258–266. [Google Scholar] [CrossRef] [Green Version]
- Jennings, L.J.; Arcila, M.E.; Corless, C.; Kamel-Reid, S.; Lubin, I.M.; Pfeifer, J.; Temple-Smolkin, R.L.; Voelkerding, K.V.; Nikiforova, M.N. Guidelines for validation of next-generation sequencing–based oncology panels. J. Mol. Diagn. 2017, 19, 341–365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strom, S.P. Current practices and guidelines for clinical next-generation sequencing oncology testing. Cancer Biol. Med. 2016, 13, 3–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torphy, R.J.; Wang, Z.; True-Yasaki, A.; Volmar, K.E.; Rashid, N.; Yeh, B. Stromal content is correlated with tissue site, contrast retention, and survival in pancreatic adenocarcinoma. JCO Precis. Oncol. 2018, 2018, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eisen, A.; Blackmore, K.M.; Meschino, W.S.; Muradali, D.; Carroll, J.C.; Majpruz, V.; Warner, E.; Rabeneck, L.; Chiarelli, A.M. Genetic assessment wait time indicators in the high risk ontario breast screening program. Mol. Genet. Genom. Med. 2018, 6, 213–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yurgelun, M.B.; Chittenden, A.B.; Ukaegbu, C.I.; Perez, K.; Madigan, A.P.; Gaonkar, S.; Cleary, J.M.; Aguirre, A.; Wolpin, B.M.; Syngal, S. Implementation of systematic genetic counseling (GC) and multigene germline testing (MGT) for pancreatic cancer (PC) patients (pts). J. Clin. Oncol. 2020, 38, 678. [Google Scholar] [CrossRef]
- Smith, A.; Bascuñana, C.; Hall, A.; Salman, A.; Andrei, A.; Volenik, A.; Rothenmund, H.; Ferland, D.; Lamoussenery, D.; Kamath, A.; et al. Establishing a clinic-based pancreatic cancer and periampullary tumour research registry in Quebec. Curr. Oncol. 2015, 22, 113–121. [Google Scholar] [CrossRef] [Green Version]
- Symecko, H.; Mueller, R.; Spielman, K.; Batson, M.; Pundock, S.; Hamilton, J.G.; Robson, M.E.; Domchek, S.M. Ten-fold increase in genetic testing in pancreatic and metastatic prostate cancer with implementation of point of care (POC) testing. J. Clin. Oncol. 2019, 37, 1506. [Google Scholar] [CrossRef]
- Sie, A.S.; Van Zelst-Stams, W.A.G.; Spruijt, L.; Mensenkamp, A.; Ligtenberg, M.J.L.; Brunner, H.G.; Prins, J.B.; Hoogerbrugge, N. More breast cancer patients prefer BRCA-mutation testing without prior face-to-face genetic counseling. Fam. Cancer 2013, 13, 143–151. [Google Scholar] [CrossRef]
- Percival, N.; George, A.; Gyertson, J.; Hamill, M.; Fernandes, A.; Davies, E.; Rahman, N.; Banerjee, S. The integration of BRCA testing into oncology clinics. Br. J. Nurs. 2016, 25, 690–694. [Google Scholar] [CrossRef]
- Rolfo, C.D.; Manca, P.; Salgado, R.; Van Dam, P.A.; Dendooven, A.; Coelho, A.M.; Gandia, J.F.; Rutten, A.; Lybaert, W.; Vermeij, J.; et al. Multidisciplinary molecular tumour board: A tool to improve clinical practice and selection accrual for clinical trials in patients with cancer. ESMO Open 2018, 3, e000398. [Google Scholar] [CrossRef] [Green Version]
- Van Der Velden, D.; Van Herpen, C.; Van Laarhoven, H.; Smit, E.; Groen, H.; Willems, S.; Nederlof, P.; Langenberg, M.; Cuppen, E.; Sleijfer, S.; et al. Molecular Tumor Boards: Current practice and future needs. Ann. Oncol. 2017, 28, 3070–3075. [Google Scholar] [CrossRef] [PubMed]
- Knepper, T.C.; Bell, G.C.; Hicks, J.K.; Padron, E.; Teer, J.K.; Vo, T.T. Key Lessons learned from moffitt’s molecular tumor board: The clinical genomics action committee experience. Oncologist 2017, 22, 144–1451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, N.M.; Michelini, V.V.; Snell, J.M.; Balu, S.; Hoyle, A.P.; Parker, J.S.; Hayward, M.C.; Eberhard, D.A.; Salazar, A.H.; McNeillie, P.; et al. Enhancing next-generation sequencing-guided cancer care through cognitive computing. Oncologist 2017, 23, 179–185. [Google Scholar] [CrossRef] [Green Version]
- Meric-Bernstam, F.; Brusco, L.; Daniels, M.S.; Wathoo, C.; Bailey, A.M.; Strong, L.; Shaw, K.; Lu, K.; Qi, Y.; Zhao, H.; et al. Incidental germline variants in 1000 advanced cancers on a prospective somatic genomic profiling protocol. Ann. Oncol. 2016, 27, 795–800. [Google Scholar] [CrossRef] [Green Version]
- Schrader, K.A.; Cheng, D.T.; Joseph, V.; Prasad, M.; Walsh, M.; Zehir, A.; Ni, A.; Thomas, T.; Benayed, R.; Ashraf, A.; et al. Germline variants in targeted tumor sequencing using matched normal DNA. JAMA Oncol. 2016, 2, 104–111. [Google Scholar] [CrossRef]
- Lawler, M.; Siu, L.L.; Rehm, H.L.; Chanock, S.J.; Alterovitz, G.; Burn, J. All the world’s a stage: Facilitating discovery science and improved cancer care through the global alliance for genomics and health. Cancer Discov. 2015, 5, 1133–1136. [Google Scholar] [CrossRef] [Green Version]
- Li, M.M.; Datto, M.; Duncavage, E.J.; Kulkarni, S.; Lindeman, N.I.; Roy, S.; Tsimberidou, A.; Vnencak-Jones, C.L.; Wolff, D.J.; Younes, A.; et al. Standards and guidelines for the interpretation and reporting of sequence variants in cancer: A joint consensus recommendation of the association for molecular pathology, american society of clinical oncology, and college of american pathologists. J. Mol. Diagn. 2017, 19, 4–23. [Google Scholar] [CrossRef] [Green Version]
- Lowery, M.A.; Jordan, E.J.; Basturk, O.; Ptashkin, R.N.; Zehir, A.; Berger, M.; Leach, T.; Herbst, B.; Askan, G.; Maynard, H.; et al. Real-time genomic profiling of pancreatic ductal Adenocarcinoma: Potential actionability and correlation with clinical phenotype. Clin. Cancer Res. 2017, 23, 6094–6100. [Google Scholar] [CrossRef] [Green Version]
- Pishvaian, M.J.; Blais, E.M.; Brody, J.R.; Lyons, E.; DeArbeloa, P.; Hendifar, A.; Mikhail, S.; Chung, V.; Sahai, V.; Sohal, D.P.S.; et al. Overall survival in patients with pancreatic cancer receiving matched therapies following molecular profiling: A retrospective analysis of the Know Your Tumor registry trial. Lancet Oncol. 2020, 21, 508–518. [Google Scholar] [CrossRef]
- Flaherty, K.T.; Gray, R.; Chen, A.; Li, S.; Patton, D.; Hamilton, S.R.; Williams, P.M.; Mitchell, E.P.; Iafrate, A.J.; Sklar, J.; et al. The molecular analysis for therapy choice (NCI-MATCH) Trial: Lessons for genomic trial design. J. Natl. Cancer Inst. 2020. [Google Scholar] [CrossRef] [Green Version]
- Skamene, T.; Siu, L.L.; Renouf, D.J.; Laskin, J.J.; Bedard, P.L.; Jones, S.J.M. Canadian profiling and targeted agent utilization trial (CAPTUR/PM.1): A phase II basket precision medicine trial. J. Clin. Oncol. 2018, 36 (Suppl. 15), TPS12127. [Google Scholar] [CrossRef]
- Dreyer, S.; Jamieson, N.; Cooke, S.; Valle, J.; McKay, C.; Biankin, A.; Chang, D.K. PRECISION-Panc: The next generation therapeutic development platform for pancreatic cancer. Clin. Oncol. 2020, 32, 1–4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Penberthy, L.; Dahman, B.A.; Petkov, V.I.; DeShazo, J.P. Effort Required in eligibility screening for clinical trials. J. Oncol. Pract. 2012, 8, 365–370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Helgeson, J.; Rammage, M.; Urman, A.; Roebuck, M.C.; Coverdill, S.; Pomerleau, K.; Dankwa-Mullan, I.; Liu, L.I.; Sweetman, R.W.; Chau, Q.; et al. Clinical performance pilot using cognitive computing for clinical trial matching at Mayo Clinic. J. Clin. Oncol. 2018, 36, e18598. [Google Scholar] [CrossRef]
- Pereira, M.A.; Chio, I.I.C. Metastasis in pancreatic ductal adenocarcinoma: Current standing and methodologies. Genes 2019, 11, 6. [Google Scholar] [CrossRef] [Green Version]
- Tiriac, H.; Belleau, P.; Engle, D.D.; Plenker, D.; Deschênes, A.; Somerville, T.D.D.; Froeling, F.E.M.; Burkhart, R.A.; Denroche, R.E.; Jang, G.H.; et al. Organoid profiling identifies common responders to chemotherapy in pancreatic cancer. Cancer Discov. 2018, 8, 1112–1129. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
| Year | Investigational Therapy | Comparator Therapy | Overall Survival (Months) | |
|---|---|---|---|---|
| Burris et al. [5] | 1997 | Gemcitabine | Fluorouracil | 5.65 vs. 4.41 |
| Moore et al. [6] NCIC CTG PA.3 | 2007 | Gemcitabine + Erlotinib * | Gemcitabine + placebo | 6.24 vs. 5.91 |
| Conroy et al. [3] PRODIGE 4/ACCORD 11 | 2011 | FOLFIRINOX | Gemcitabine | 11.1 vs. 6.8 |
| Von Hoff et al. [4] MPACT | 2013 | Gemcitabine + Nab-Paclitaxel | Gemcitabine | 8.5 vs. 6.7 |
| Wang-Gillam et al. [7] NAPOLI-1 | 2016 | Nanoliposomal irinotecan + fluorouracil + folinic acid | Fluorouracil + folinic acid | 6.1 vs. 4.2 |
| Year | Investigational Therapy | Comparator Therapy | Overall Survival (Months) | |
|---|---|---|---|---|
| Neoptolemos et al. [8] ESPAC-1 | 2004 | Fluorouracil | No adjuvant therapy | 20.1 vs. 15.5 |
| Oettle et al. [9] CONKO-001 | 2013 | Gemcitabine | No adjuvant therapy | 22.8 vs. 20.2 |
| Neoptolemos et al. [10] ESPAC-4 | 2017 | Gemcitabine + Capecitabine | Gemcitabine | 28.0 vs. 25.5 |
| Conroy et al. [11] NCIC CTG PA.6 | 2018 | mFOLFIRINOX | Gemcitabine | 54.4 vs. 35.0 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.; Lakoma, A.; Zogopoulos, G. Building towards Precision Oncology for Pancreatic Cancer: Real-World Challenges and Opportunities. Genes 2020, 11, 1098. https://doi.org/10.3390/genes11091098
Wang Y, Lakoma A, Zogopoulos G. Building towards Precision Oncology for Pancreatic Cancer: Real-World Challenges and Opportunities. Genes. 2020; 11(9):1098. https://doi.org/10.3390/genes11091098
Chicago/Turabian StyleWang, Yifan, Anna Lakoma, and George Zogopoulos. 2020. "Building towards Precision Oncology for Pancreatic Cancer: Real-World Challenges and Opportunities" Genes 11, no. 9: 1098. https://doi.org/10.3390/genes11091098