Two Cases of Recessive Intellectual Disability Caused by NDST1 and METTL23 Variants

 , ,
, ,  , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Subjects
2.2. DNA Extraction and Quantification
2.3. Library Construction and Exome Sequencing
2.4. Data Processing
2.5. Variant Filtration Steps
2.6. Primer Design and Variant Confirmation
2.7. Homology Protein Modeling
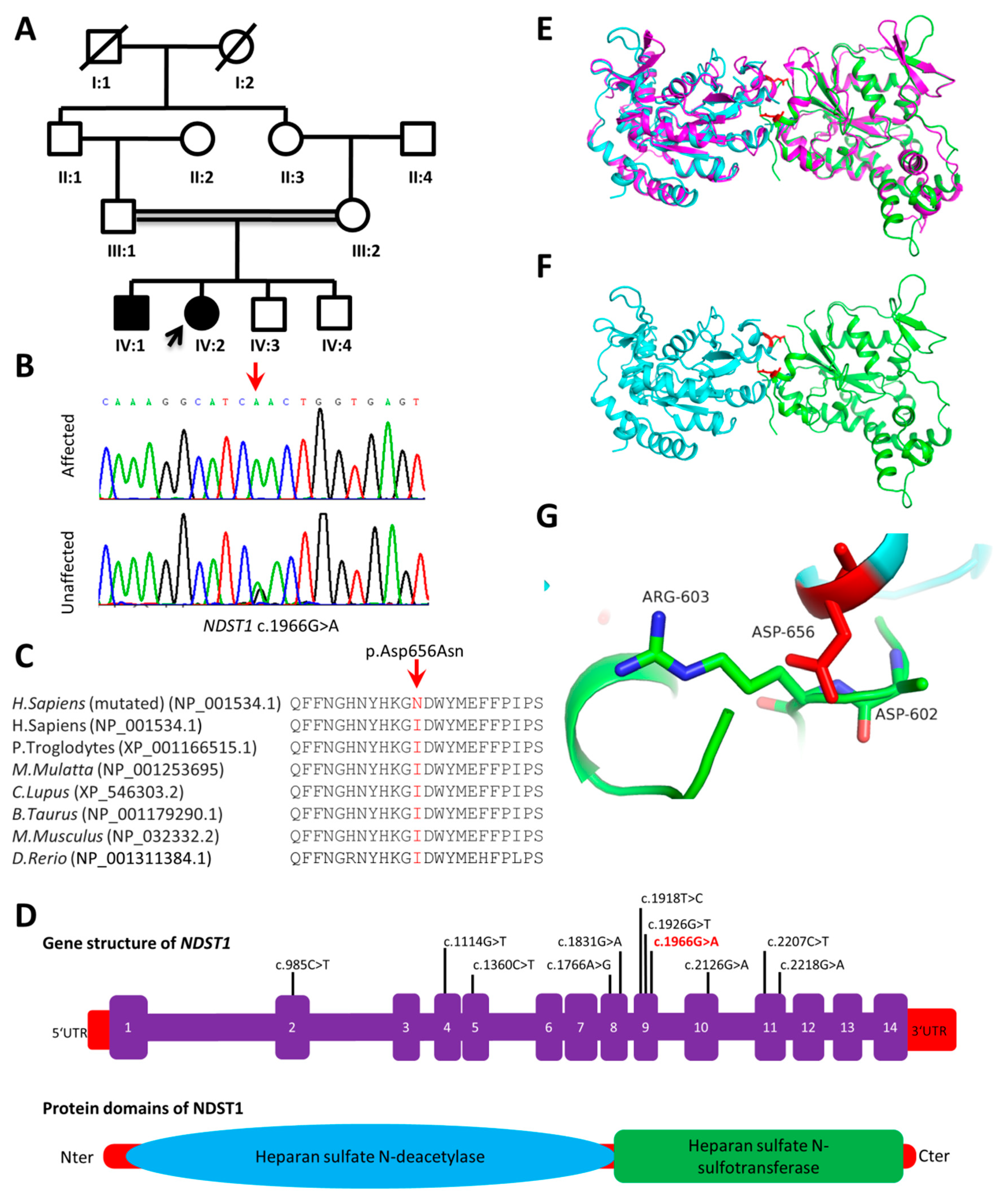
2.7.1. NDST1 (p.Asp656Asn)
2.7.2. METTL23 (p.Phe104Leu)
3. Results
3.1. Clinical Manifestations
3.1.1. Family A
3.1.2. Family B
3.2. Molecular Evaluation
3.3. Homology Modeling
3.3.1. NDST1 (p.Asp656Asn)
3.3.2. METTL23 (p.Phe104Leu)
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Musante, L.; Ropers, H.-H. Genetics of recessive cognitive disorders. Trends Genet. 2014, 30, 32–39. [Google Scholar] [CrossRef] [Green Version]
- Maulik, P.K.; Mascarenhas, M.N.; Mathers, C.D.; Dua, T.; Saxena, S. Prevalence of intellectual disability: A meta-analysis of population-based studies. Res. Dev. Disabil. 2011, 32, 419–436. [Google Scholar] [CrossRef]
- Moeschler, J.B.; Shevell, M. Committee on Genetics Comprehensive Evaluation of the Child with Intellectual Disability or Global Developmental Delays. Pediatrics 2014, 134, e903–e918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McRae, J.F.; Clayton, S.; Fitzgerald, T.W.; Kaplanis, J.; Prigmore, E.; Rajan, D.; Sifrim, A.; Aitken, S.; Akawi, N.; Alvi, M.; et al. Prevalence and architecture of de novo mutations in developmental disorders. Nature 2017, 542, 433–438. [Google Scholar] [CrossRef]
- Rauch, A.; Hoyer, J.; Guth, S.; Zweier, C.; Kraus, C.; Becker, C.; Zenker, M.; Hüffmeier, U.; Thiel, C.T.; Rüschendorf, F.; et al. Diagnostic yield of various genetic approaches in patients with unexplained developmental delay or mental retardation. Am. J. Med. Genet. Part A 2006, 140, 2063–2074. [Google Scholar] [CrossRef]
- Reuter, M.S.; Tawamie, H.; Buchert, R.; Gebril, O.H.; Froukh, T.; Thiel, C.T.; Uebe, S.; Ekici, A.B.; Krumbiegel, M.; Zweier, C.; et al. Diagnostic Yield and Novel Candidate Genes by Exome Sequencing in 152 Consanguineous Families With Neurodevelopmental Disorders. JAMA Psychiatry 2017, 74, 293–299. [Google Scholar] [CrossRef] [PubMed]
- Gilissen, C.; Hehir-Kwa, J.Y.; Thung, D.T.; Van De Vorst, M.; Van Bon, B.W.; Willemsen, M.H.; Kwint, M.; Janssen, I.M.; Hoischen, A.; Schenck, A.; et al. Genome sequencing identifies major causes of severe intellectual disability. Nature 2014, 511, 344–347. [Google Scholar] [CrossRef] [PubMed]
- De Ligt, J.; Willemsen, M.H.; Van Bon, B.W.; Kleefstra, T.; Yntema, H.G.; Kroes, T.; Silfhout, A.T.V.-V.; Koolen, D.A.; De Vries, P.; Gilissen, C.; et al. Diagnostic Exome Sequencing in Persons with Severe Intellectual Disability. N. Engl. J. Med. 2012, 367, 1921–1929. [Google Scholar] [CrossRef] [Green Version]
- Ropers, H.-H. Genetics of Early Onset Cognitive Impairment. Annu. Rev. Genom. Hum. Genet. 2010, 11, 161–187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fitzgerald, T.W.; Gerety, S.S.; Jones, W.D.; van Kogelenberg, M.; King, D.A.; McRae, J.; Morley, K.; Parthiban, V.; Al-Turki, S.; Ambridge, K.; et al. Large-scale discovery of novel genetic causes of developmental disorders. Nature 2014, 519, 223–228. [Google Scholar] [CrossRef]
- Wright, C.F.; Fitzgerald, T.; Jones, W.D.; Clayton, S.; McRae, J.F.; Van Kogelenberg, M.; King, D.A.; Ambridge, K.; Barrett, D.M.; Bayzetinova, T.; et al. Genetic diagnosis of developmental disorders in the DDD study: A scalable analysis of genome-wide research data. Lancet 2015, 385, 1305–1314. [Google Scholar] [CrossRef] [Green Version]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–423. [Google Scholar] [CrossRef] [PubMed]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; Beer, T.A.P.D.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erzurumluoglu, A.M.; Shihab, H.A.; Rodriguez, S.; Gaunt, T.R.; Day, I.N. Importance of Genetic Studies in Consanguineous Populations for the Characterization of Novel Human Gene Functions. Ann. Hum. Genet. 2016, 80, 187–196. [Google Scholar] [CrossRef] [Green Version]
- Reuter, M.S.; Musante, L.; Hu, H.; Diederich, S.; Sticht, H.; Ekici, A.B.; Uebe, S.; Wienker, T.F.; Bartsch, O.; Zechner, U.; et al. NDST1missense mutations in autosomal recessive intellectual disability. Am. J. Med. Genet. Part A 2014, 164, 2753–2763. [Google Scholar] [CrossRef] [Green Version]
- Bernkopf, M.; Webersinke, G.; Tongsook, C.; Koyani, C.; Rafiq, M.A.; Ayaz, M.; Müller, D.; Enzinger, C.; Aslam, M.; Naeem, F.; et al. Disruption of the methyltransferase-like 23 gene METTL23 causes mild autosomal recessive intellectual disability. Hum. Mol. Genet. 2014, 23, 4015–4023. [Google Scholar] [CrossRef] [Green Version]
- Reiff, R.E.; Ali, B.R.S.; Baron, B.; Yu, T.W.; Ben-Salem, S.; Coulter, M.E.; Schubert, C.R.; Hill, R.S.; Akawi, N.A.; Al-Younes, B.; et al. METTL23, a transcriptional partner of GABPA, is essential for human cognition. Hum. Mol. Genet. 2014, 23, 3456–3466. [Google Scholar] [CrossRef]
- Filipek-Gorniok, B.; Carlsson, P.; Haitina, T.; Habicher, J.; Ledin, J.; Kjellén, L. The Ndst Gene Family in Zebrafish: Role of Ndst1b in Pharyngeal Arch Formation. PLoS ONE 2015, 10, e0119040. [Google Scholar] [CrossRef] [Green Version]
- Grobe, K.; Inatani, M.; Pallerla, S.R.; Castagnola, J.; Yamaguchi, Y.; Esko, J.D. Cerebral hypoplasia and craniofacial defects in mice lacking heparan sulfate Ndst1 gene function. Development 2005, 132, 3777–3786. [Google Scholar] [CrossRef] [Green Version]
- Almannai, M.; Obaid, O.; Faqeih, E.; Alasmari, A.; Samman, M.M.; Pinz, H.; Braddock, S.R.; Alkuraya, F.S. Further delineation of METTL23 -associated intellectual disability. Am. J. Med. Genet. Part A 2020, 182, 785–791. [Google Scholar] [CrossRef]
- Smaili, W.; Elalaoui, S.C.; Zrhidri, A.; Raymond, L.; Egéa, G.; Taoudi, M.; Mouatassim, S.; Sefiani, A.; Lyahyai, J. Exome sequencing revealed a novel homozygous METTL23 gene mutation leading to familial mild intellectual disability with dysmorphic features. Eur. J. Med. Genet. 2020, 63, 103951. [Google Scholar] [CrossRef] [PubMed]
- Harripaul, R.; Vasli, N.; Mikhailov, A.; Rafiq, M.A.; Mittal, K.; Windpassinger, C.; Sheikh, T.I.; Noor, A.; Mahmood, H.; Downey, S.; et al. Mapping autosomal recessive intellectual disability: Combined microarray and exome sequencing identifies 26 novel candidate genes in 192 consanguineous families. Mol. Psychiatry 2017, 23, 973–984. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Clinical Findings | Family A | Family B | |||
|---|---|---|---|---|---|
| Sex | M | F | F | F | F |
| Age (years) | 12 | 10 | 24 | 12 | 10 |
| Developmental delay | + | + | + | + | + |
| Intellectual disability | ++ | ++ | ++ | ++ | + |
| Microcephaly | - | - | - | - | - |
| Hypothyroidism | - | - | - | - | - |
| Speech development delay | + | + | - | - | - |
| Learning disability | + | + | + | + | + |
| Behavioral abnormalities | + | + | + | + | + |
| Dysmorphic features | - | - | + | + | - |
| Skeletal problem | - | - | - | - | - |
| Ophthalmic problem | - | - | - | - | - |
| Skin problem | - | - | - | - | - |
| Seizures | + | + | + | + | + |
| Social activity | + | + | + | + | - |
| Muscular dystrophy | - | - | - | - | - |
| Self biting | - | - | ++ | ++ | + |
| Family ID | Family A | Family B |
|---|---|---|
| Affected individuals | IV-1, IV-2 | V-2, V-4, V-5 |
| Transcript ID | NM_001543.5 | NM_001206984.3 |
| Gene name | NDST1 | METTL23 |
| MIM number | 616116 | 615942 |
| Chromosome position | Chr5:149922529 | Chr17:74729285 |
| Nucleotide change | c.1966G>A | c.310T>C |
| Protein change | p.Asp656Asn | p.Phe104Leu |
| SNP number | rs150320391 | - |
| GnomAD frequency | 0.000016 | - |
| SIFT score | 0.08/T | 0/D |
| Polyphen2 score | 0.992/D | 0.891/PD |
| Mutation taster score | 1/D | 1/D |
| CADD score | 27.4 | 31 |
| PROVEAN score | −0.17/N | −4.67/D |
| ACMG classification | PM1 | PM2 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khan, A.; Miao, Z.; Umair, M.; Ullah, A.; Alshabeeb, M.A.; Bilal, M.; Ahmad, F.; Rappold, G.A.; Ansar, M.; Carapito, R. Two Cases of Recessive Intellectual Disability Caused by NDST1 and METTL23 Variants. Genes 2020, 11, 1021. https://doi.org/10.3390/genes11091021
Khan A, Miao Z, Umair M, Ullah A, Alshabeeb MA, Bilal M, Ahmad F, Rappold GA, Ansar M, Carapito R. Two Cases of Recessive Intellectual Disability Caused by NDST1 and METTL23 Variants. Genes. 2020; 11(9):1021. https://doi.org/10.3390/genes11091021
Chicago/Turabian StyleKhan, Amjad, Zhichao Miao, Muhammad Umair, Amir Ullah, Mohammad A. Alshabeeb, Muhammad Bilal, Farooq Ahmad, Gudrun A. Rappold, Muhammad Ansar, and Raphael Carapito. 2020. "Two Cases of Recessive Intellectual Disability Caused by NDST1 and METTL23 Variants" Genes 11, no. 9: 1021. https://doi.org/10.3390/genes11091021