Comparative Analysis of the Transcriptome and Distribution of Putative SNPs in Two Rainbow Trout (Oncorhynchus mykiss) Breeding Strains by Using Next-Generation Sequencing

 ,
,  ,
,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Experimental Animals
2.2. Nucleotide Extraction and Library Preparation
2.3. Sequencing Data Processing and Analysis of Differential Expression
2.4. Identification of Putative SNPs
2.5. Validation of Putative SNPs by Resequencing
2.6. Data Deposition
2.7. Ethical Statement
3. Results and Discussion
3.1. A Total of 1760 Annotated Genes Were Differently Expressed Between Rainbow Trout Strains Silver Steelhead and Born
3.2. TP53 Plays a Prominent Role as Upstream Regulator of Different Gene Expression Patterns in Both Trout Strains
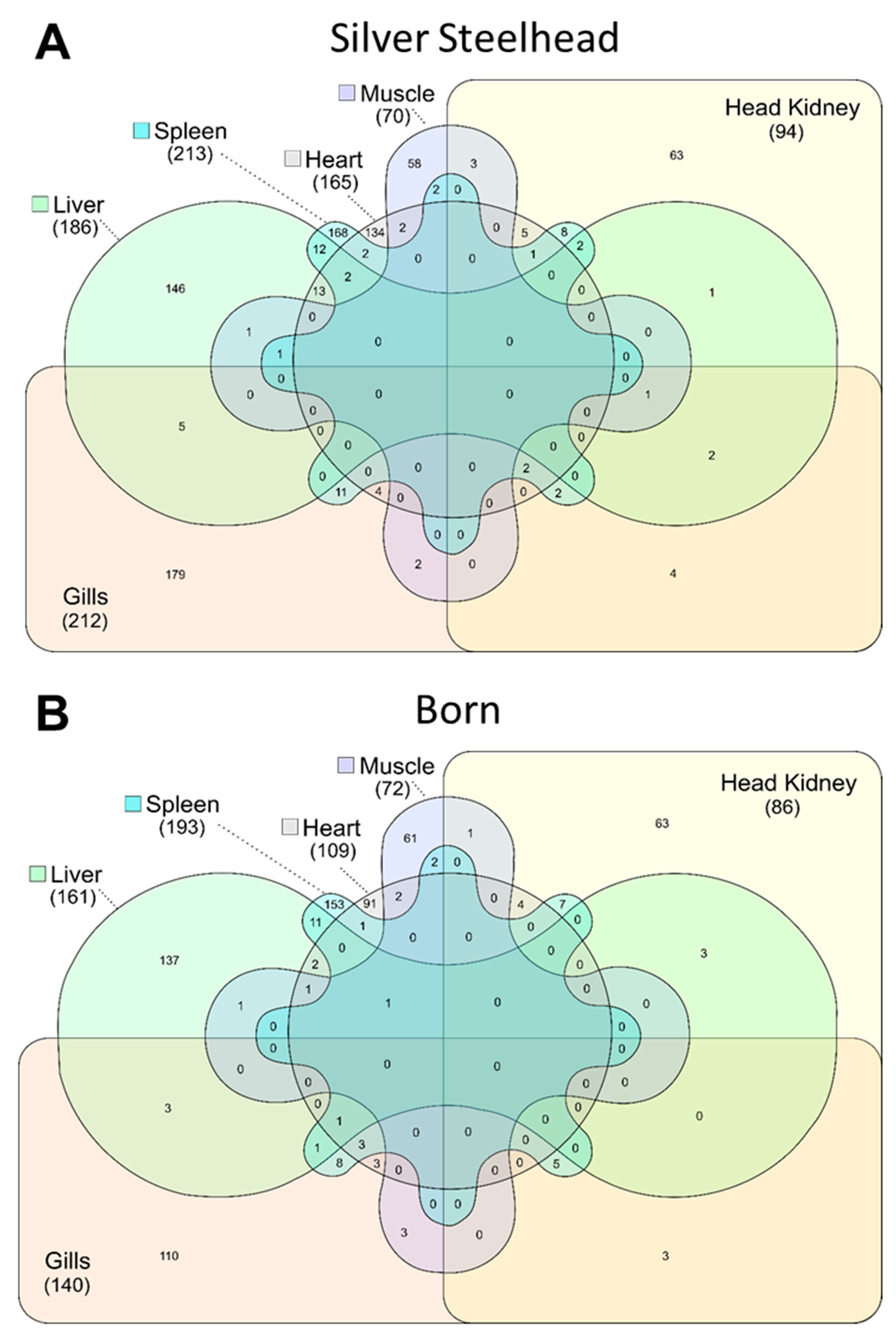
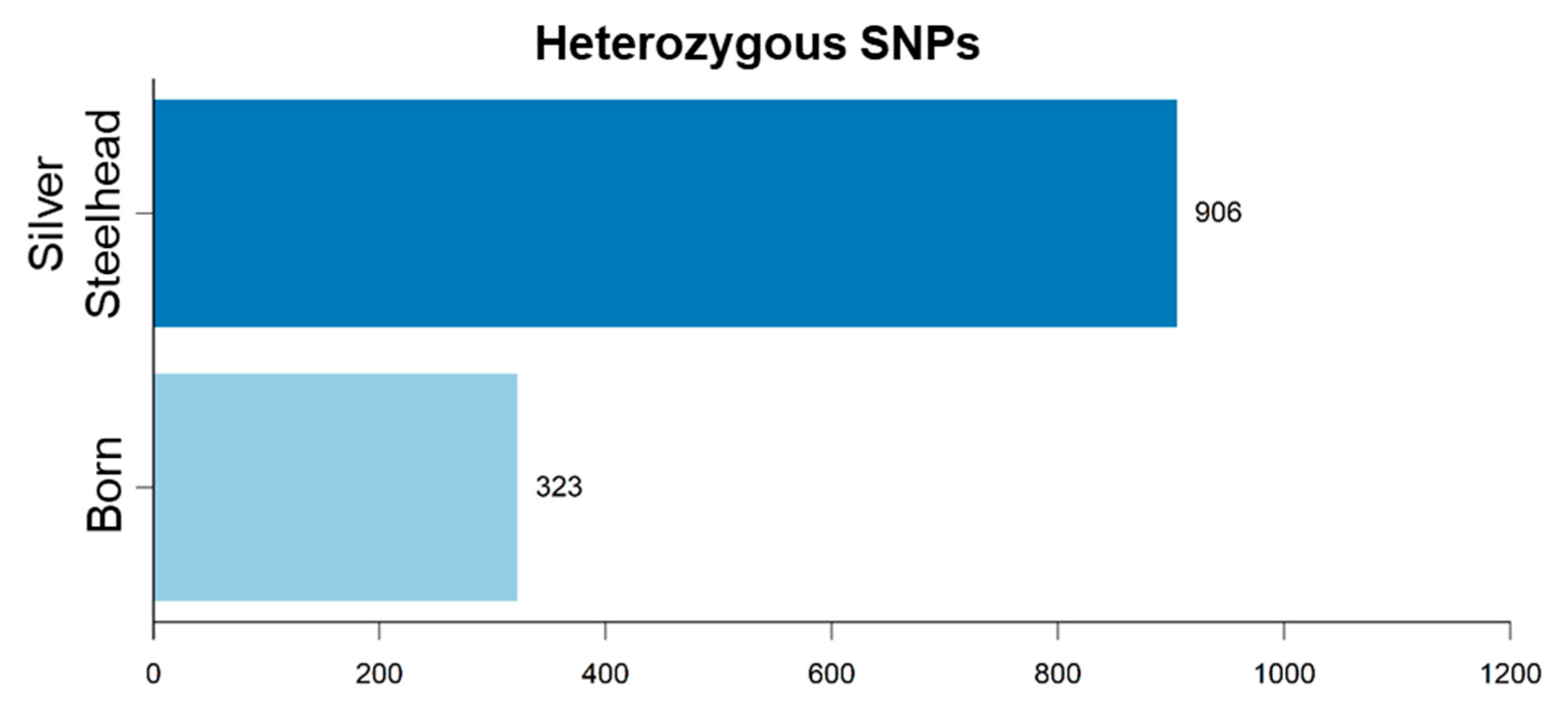
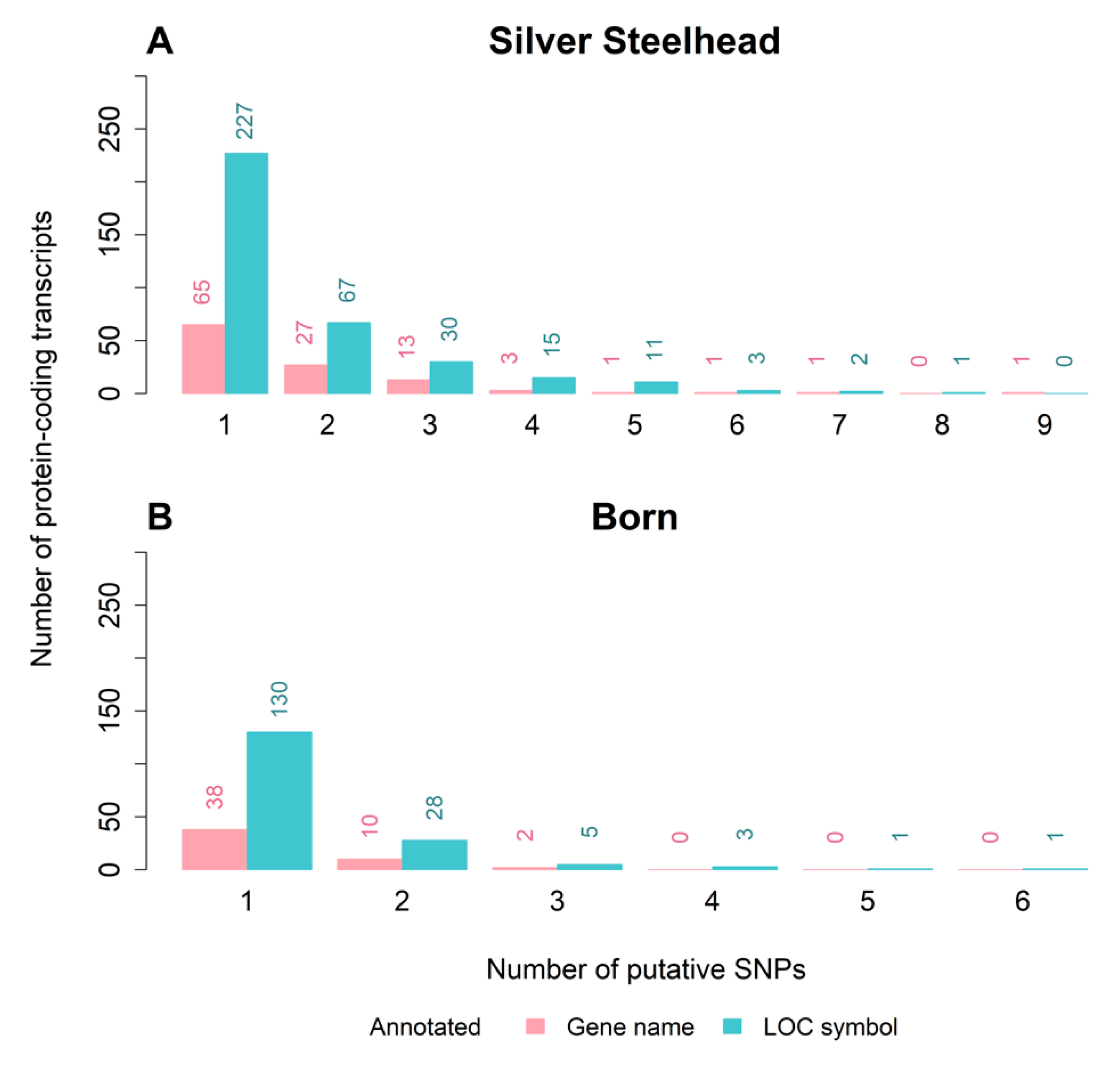
3.3. Strain-Specific SNPs Were Identified in Silver Steelhead and Born Trout
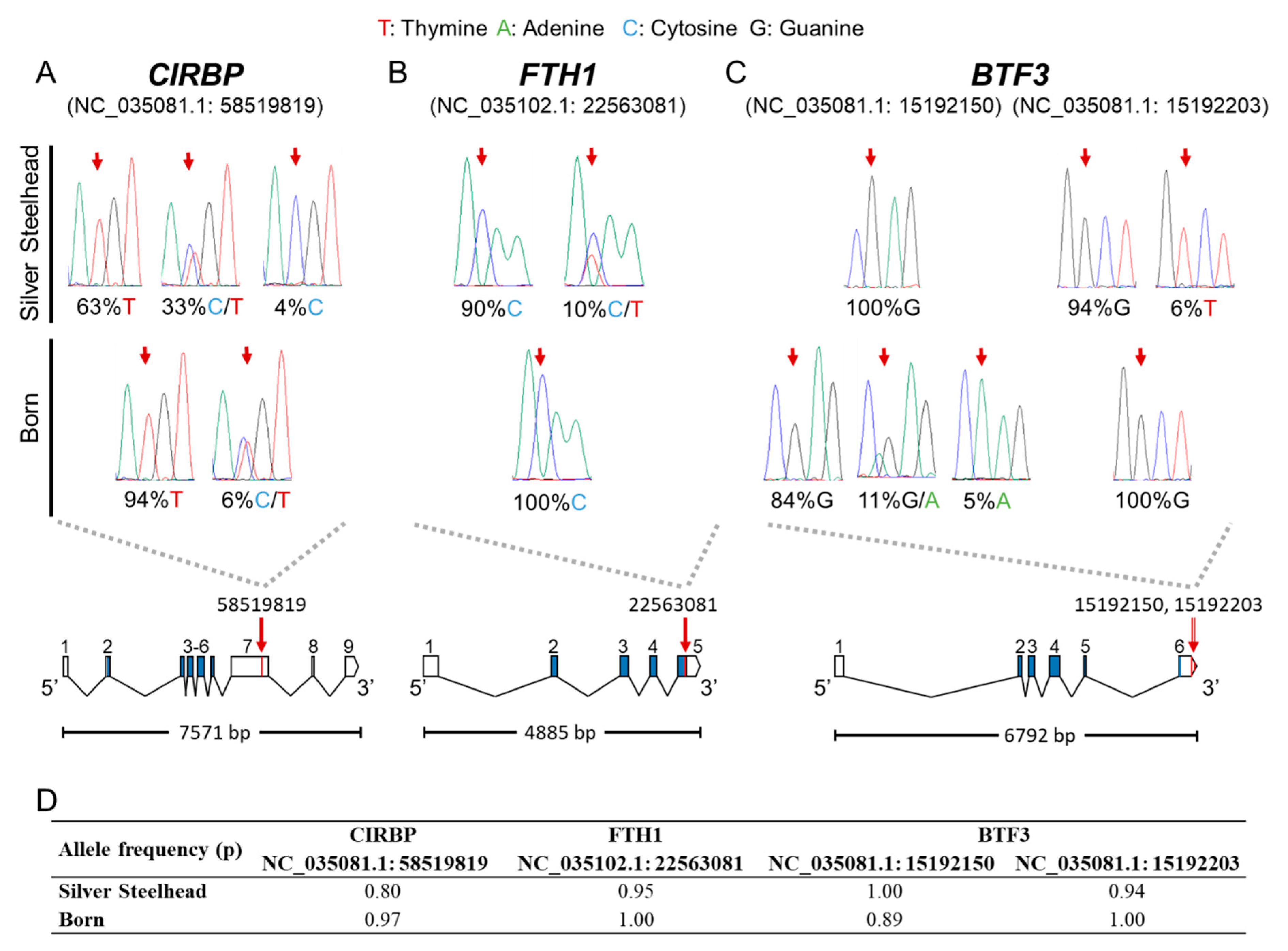
3.4. Putative SNPs Were Validated by Resequencing
3.5. Do the Identified Expression Differences and Genetic Variances Reflect a Specific Adaptation of the Born Strain?
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- National Marine Fisheries Service. Fisheries of the United States, 2016. 2017. Available online: https://www.st.nmfs.noaa.gov/commercial-fisheries/fus/fus16/index (accessed on 10 February 2020).
- FAO. The State of World Fisheries and Aquaculture 2020; FAO: Rome, Italy, 2020; ISBN 978-92-5-132692. [Google Scholar]
- Gross, R.; Lulla, P.; Paaver, T. Genetic variability and differentiation of rainbow trout (Oncorhynchus mykiss) strains in northern and Eastern Europe. Aquaculture 2007, 272, S139–S146. [Google Scholar] [CrossRef]
- Okumuş, İ. Rainbow trout Broodstock Management and Seed Production in Turkey: Present Practices, Constrains and the Future. Turk. J. Fish. Aquat. Sc. 2002, 2, 41–56. [Google Scholar]
- Gjedrem, T. Genetic improvement of cold-water fish species. Aquac. Res. 2000, 31, 25–33. [Google Scholar] [CrossRef]
- Jones, M.J.; Fejes, A.P.; Kobor, M.S. DNA methylation, genotype and gene expression: Who is driving and who is along for the ride? Genome Boil. 2013, 14, 126. [Google Scholar] [CrossRef] [PubMed]
- Moghadam, H.; Mørkøre, T.; Robinson, N. Epigenetics—Potential for Programming Fish for Aquaculture? J. Mar. Sci. Eng. 2015, 3, 175–192. [Google Scholar] [CrossRef] [Green Version]
- Lim, I.Y.; Lin, X.; Karnani, N. Implications of Genotype and Environment on Variation in DNA Methylation. In Handbook of Nutrition, Diet, and Epigenetics; Springer Science and Business Media LLC: New York, NY, USA, 2017; pp. 1–20. [Google Scholar]
- Anders, E. Stand der Züchtung und Reproduktion brackwasseradaptierter Regenbogenforellenbestände im Küstenbereich der DDR. Fischerei-Forschung 1986, 24, 72. [Google Scholar]
- Rebl, A.; Verleih, M.; Korytář, T.; Kühn, C.; Wimmers, K.; Köllner, B.; Goldammer, T. Identification of differentially expressed protective genes in liver of two rainbow trout strains. Veter-Immunol. Immunopathol. 2012, 145, 305–315. [Google Scholar] [CrossRef]
- Borchel, A.; Verleih, M.; Rebl, A.; Goldammer, T. Identification of genes involved in cold-shock response in rainbow trout (Oncorhynchus mykiss). J. Genet. 2017, 96, 701–706. [Google Scholar] [CrossRef]
- Köbis, J.M.; Rebl, A.; Kühn, C.; Goldammer, T. Comparison of splenic transcriptome activity of two rainbow trout strains differing in robustness under regional aquaculture conditions. Mol. Biol. Rep. 2012, 40, 1955–1966. [Google Scholar] [CrossRef]
- Rebl, A.; Verleih, M.; Köbis, J.M.; Kühn, C.; Wimmers, K.; Köllner, B.; Goldammer, T. Transcriptome Profiling of Gill Tissue in Regionally Bred and Globally Farmed Rainbow Trout Strains Reveals Different Strategies for Coping with Thermal Stress. Mar. Biotechnol. 2013, 15, 445–460. [Google Scholar] [CrossRef]
- Verleih, M.; Borchel, A.; Krasnov, A.; Rebl, A.; Korytář, T.; Kühn, C.; Goldammer, T. Impact of Thermal Stress on Kidney-Specific Gene Expression in Farmed Regional and Imported Rainbow Trout. Mar. Biotechnol. 2015, 17, 576–592. [Google Scholar] [CrossRef] [PubMed]
- Rebl, A.; Korytář, T.; Köbis, J.M.; Verleih, M.; Krasnov, A.; Jaros, J.; Kühn, C.; Köllner, B.; Goldammer, T. Transcriptome Profiling Reveals Insight into Distinct Immune Responses to Aeromonas salmonicida in Gill of Two Rainbow Trout Strains. Mar. Biotechnol. 2013, 16, 333–348. [Google Scholar] [CrossRef] [PubMed]
- Köbis, J.M.; Rebl, A.; Kühn, C.; Korytář, T.; Köllner, B.; Goldammer, T. Comprehensive and comparative transcription analyses of the complement pathway in rainbow trout. Fish Shellfish. Immunol. 2015, 42, 98–107. [Google Scholar] [CrossRef] [PubMed]
- Gjedrem, T.; Robinson, N.; Rye, M.; Robinson, N.A. The importance of selective breeding in aquaculture to meet future demands for animal protein: A review. Aquaculture 2012, 350, 117–129. [Google Scholar] [CrossRef]
- Glover, K.A. Genetic characterisation of farmed rainbow trout in Norway: Intra- and inter-strain variation reveals potential for identification of escapees. BMC Genet. 2008, 9, 87. [Google Scholar] [CrossRef] [Green Version]
- Abdelrahman, H.; Genomics, G.A.B.W.T.A.; Elhady, M.; Alcivar-Warren, A.; Allen, S.; Al-Tobasei, R.; Bao, L.; Beck, B.; Blackburn, H.; Bosworth, B.; et al. Aquaculture genomics, genetics and breeding in the United States: Current status, challenges, and priorities for future research. BMC Genom. 2017, 18, 1–23. [Google Scholar] [CrossRef] [Green Version]
- Tan, E.; Kinoshita, S.; Suzuki, Y.; Ineno, T.; Tamaki, K.; Kera, A.; Muto, K.; Yada, T.; Kitamura, S.; Asakawa, S.; et al. Different gene expression profiles between normal and thermally selected strains of rainbow trout, Oncorhynchus mykiss, as revealed by comprehensive transcriptome analysis. Gene 2016, 576, 637–643. [Google Scholar] [CrossRef]
- Danzmann, R.G.; Kocmarek, A.L.; Norman, J.D.; Rexroad, C.E.; Palti, Y. Transcriptome profiling in fast versus slow-growing rainbow trout across seasonal gradients. BMC Genom. 2016, 17, 60. [Google Scholar] [CrossRef] [Green Version]
- Ali, A.; Al-Tobasei, R.; Kenney, B.; Leeds, T.D.; Salem, M. Integrated analysis of lncRNA and mRNA expression in rainbow trout families showing variation in muscle growth and fillet quality traits. Sci. Rep. 2018, 8, 12111. [Google Scholar] [CrossRef]
- Al-Tobasei, R.; Ali, A.; Leeds, T.D.; Liu, S.; Palti, Y.; Kenney, P.B.; Salem, M. Identification of SNPs associated with muscle yield and quality traits using allelic-imbalance analyses of pooled RNA-Seq samples in rainbow trout. BMC Genom. 2017, 18, 1–15. [Google Scholar] [CrossRef]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. 2010. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc (accessed on 20 May 2019).
- Gao, G.; Nome, T.; Pearse, D.E.; Moen, T.; Naish, K.-A.; Thorgaard, G.H.; Lien, S.; Palti, Y. A New Single Nucleotide Polymorphism Database for Rainbow Trout Generated Through Whole Genome Resequencing. Front. Genet. 2018, 9, 147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.; Langmead, B.; Salzberg, S. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.-C.; Mendell, J.T.; Salzberg, S. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Boil. 2014, 15, 31. [Google Scholar] [CrossRef] [Green Version]
- Van Der Auwera, G.A.; O Carneiro, M.; Hartl, C.; Poplin, R.; Del Angel, G.; Levy-Moonshine, A.; Jordan, T.; Shakir, K.; Roazen, D.; Thibault, J.; et al. From FastQ Data to High-Confidence Variant Calls: The Genome Analysis Toolkit Best Practices Pipeline. Curr. Protoc. Bioinform. 2013, 43, 11.10.1–11.10.33. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2012, 29, 15–21. [Google Scholar] [CrossRef]
- Kapitonov, V.V.; Jurka, J. HarbingerTransposons and an Ancient HARBI1 Gene Derived from a Transposase. DNA Cell Boil. 2004, 23, 311–324. [Google Scholar] [CrossRef]
- Lu, S.C. Glutathione synthesis. Biochim. Biophys. Acta (BBA) Bioenergy 2012, 1830, 3143–3153. [Google Scholar] [CrossRef] [Green Version]
- Valero, Y.; Martínez-Morcillo, F.J.; Esteban, M. Ángeles; Chaves-Pozo, E.; Cuesta, A. Fish Peroxiredoxins and Their Role in Immunity. Biology 2015, 4, 860–880. [Google Scholar] [CrossRef] [Green Version]
- Coutermarsh-Ott, S.; Eden, K.; Allen, I.C. Beyond the inflammasome: Regulatory NOD-like receptor modulation of the host immune response following virus exposure. J. Gen. Virol. 2016, 97, 825–838. [Google Scholar] [CrossRef] [Green Version]
- Grimsey, N.L.; Savinainen, J.R.; Attili, B.; Ahamed, M. Regulating membrane lipid levels at the synapse by small-molecule inhibitors of monoacylglycerol lipase: New developments in therapeutic and PET imaging applications. Drug Discov. Today 2020, 25, 330–343. [Google Scholar] [CrossRef]
- Glisovic, T.; Bachorik, J.L.; Yong, J.; Dreyfuss, G. RNA-binding proteins and post-transcriptional gene regulation. FEBS Lett. 2008, 582, 1977–1986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dolgin, E. The most popular genes in the human genome. Nature 2017, 551, 427–431. [Google Scholar] [CrossRef] [PubMed]
- Vousden, K.H.; Lane, D. p53 in health and disease. Nat. Rev. Mol. Cell Boil. 2007, 8, 275–283. [Google Scholar] [CrossRef] [PubMed]
- Lane, D. p53, guardian of the genome. Nature 1992, 358, 15–16. [Google Scholar] [CrossRef]
- Brady, C.A.; Attardi, L.D. p53 at a glance. J. Cell Sci. 2010, 123, 2527–2532. [Google Scholar] [CrossRef] [Green Version]
- Levine, A.J. p53, the Cellular Gatekeeper for Growth and Division. Cell 1997, 88, 323–331. [Google Scholar] [CrossRef] [Green Version]
- The 1000 Genomes Project Consortium; 1000 Genomes Project Consortium; Abecasis, G.R.; Altshuler, D.M.; Auton, A.; Brooks, L.D.; Durbin, R.M.; Gibbs, R.A.; Hurles, M.E.; McVean, G.A.; et al. A map of human genome variation from population-scale sequencing. Nature 2010, 467, 1061–1073. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Liu, S.; Yao, J.; Bao, L.; Zhang, J.; Li, Y.; Jiang, C.; Sun, L.; Wang, R.; Zhang, Y.; et al. The channel catfish genome sequence provides insights into the evolution of scale formation in teleosts. Nat. Commun. 2016, 7, 11757. [Google Scholar] [CrossRef]
- Vera, M.; Álvarez-Dios, J.A.; Fernández, J.; Bouza, C.; Vilas, R.; Portela, P.M. Development and Validation of Single Nucleotide Polymorphisms (SNPs) Markers from Two Transcriptome 454-Runs of Turbot (Scophthalmus maximus) Using High-Throughput Genotyping. Int. J. Mol. Sci. 2013, 14, 5694–5711. [Google Scholar] [CrossRef] [Green Version]
- Smith, C.T.; Elfstrom, C.M.; Seeb, L.W.; Seeb, J.E. Use of sequence data from rainbow trout and Atlantic salmon for SNP detection in Pacific salmon. Mol. Ecol. 2005, 14, 4193–4203. [Google Scholar] [CrossRef] [PubMed]
- Berthelot, C.; Brunet, F.; Chalopin, M.; Juanchich, A.; Bernard, M.; Noel, B.; Bento, P.; Da Silva, C.; Labadie, K.; Alberti, A.; et al. The rainbow trout genome provides novel insights into evolution after whole-genome duplication in vertebrates. Nat. Commun. 2014, 5, 3657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allendorf, F.W.; Thorgaard, G.H. Tetraploidy and the Evolution of Salmonid Fishes. In Evolutionary Genetics of Fishes; Springer Science and Business Media LLC: New York, NY, USA, 1984; pp. 1–53. [Google Scholar]
- Robertson, F.; Gundappa, M.K.; Grammes, F.; Hvidsten, T.R.; Redmond, A.; Lien, S.; Martin, S.A.M.; Holland, P.; Sandve, S.R.; MacQueen, D.J. Lineage-specific rediploidization is a mechanism to explain time-lags between genome duplication and evolutionary diversification. Genome Boil. 2017, 18, 111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryynänen, H.J.; Primmer, C.R. Single nucleotide polymorphism (SNP) discovery in duplicated genomes: Intron-primed exon-crossing (IPEC) as a strategy for avoiding amplification of duplicated loci in Atlantic salmon (Salmo salar) and other salmonid fishes. BMC Genom. 2006, 7, 192. [Google Scholar] [CrossRef] [Green Version]
- Fredman, D.; White, S.J.; Potter, S.; E Eichler, E.; Dunnen, J.T.D.; Brookes, A.J. Complex SNP-related sequence variation in segmental genome duplications. Nat. Genet. 2004, 36, 861–866. [Google Scholar] [CrossRef]
- Sanchez, C.C.; Palti, Y.; Rexroad, C. SNP Analysis with Duplicated Fish Genomes: Differentiation of SNPs, Paralogous Sequence Variants, and Multisite Variants. In Next Generation Sequencing and Whole Genome Selection in Aquaculture; Wiley: New York, NY, USA, 2010; pp. 133–150. [Google Scholar]
- Gidskehaug, L.; Kent, M.; Hayes, B.J.; Lien, S. Genotype calling and mapping of multisite variants using an Atlantic salmon iSelect SNP array. Bioinformatics 2010, 27, 303–310. [Google Scholar] [CrossRef] [Green Version]
- Palti, Y.; Gao, G.; Liu, S.; Kent, M.P.; Lien, S.; Miller, M.R.; Rexroad, C.E.; Moen, T. The development and characterization of a 57K single nucleotide polymorphism array for rainbow trout. Mol. Ecol. Resour. 2014, 15, 662–672. [Google Scholar] [CrossRef]
- Salem, M.; Al-Tobasei, R.; Ali, A.; Lourenco, D.; Gao, G.; Palti, Y.; Kenney, B.; Leeds, T.D. Genome-Wide Association Analysis With a 50K Transcribed Gene SNP-Chip Identifies QTL Affecting Muscle Yield in Rainbow Trout. Front. Genet. 2018, 9. [Google Scholar] [CrossRef]
- Castle, J.C. SNPs Occur in Regions with Less Genomic Sequence Conservation. PLoS ONE 2011, 6, e20660. [Google Scholar] [CrossRef]
- Ramírez-Bello, J.; Jiménez-Morales, M. [Functional implications of single nucleotide polymorphisms (SNPs) in protein-coding and non-coding RNA genes in multifactorial diseases]. Gac. Med. Mex 2017, 153, 238–250. [Google Scholar]
- Hrdlickova, B.; De Almeida, R.C.; Borek, Z.; Withoff, S. Genetic variation in the non-coding genome: Involvement of micro-RNAs and long non-coding RNAs in disease. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2014, 1842, 1910–1922. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayes, B.J.; Laerdahl, J.K.; Lien, S.; Moen, T.; Berg, P.; Hindar, K.; Davidson, W.; Koop, B.F.; Adzhubei, A.; Høyheim, B. An extensive resource of single nucleotide polymorphism markers associated with Atlantic salmon (Salmo salar) expressed sequences. Aquaculture 2007, 265, 82–90. [Google Scholar] [CrossRef]
- Hubert, S.; Higgins, B.; Borza, T.; Bowman, S. Development of a SNP resource and a genetic linkage map for Atlantic cod (Gadus morhua). BMC Genom. 2010, 11, 191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diopere, E.; Hellemans, B.; Volckaert, F.A.; Maes, G. Identification and validation of single nucleotide polymorphisms in growth- and maturation-related candidate genes in sole (Solea solea L.). Mar. Genom. 2013, 9, 33–38. [Google Scholar] [CrossRef]
- Cui, J.; Wang, H.; Liu, S.; Zhu, L.; Qiu, X.; Jiang, Z.; Wang, X.; Liu, Z. SNP Discovery from Transcriptome of the Swimbladder of Takifugu rubripes. PLoS ONE 2014, 9, e92502. [Google Scholar] [CrossRef]
- Hunt, R.; Sauna, Z.E.; Ambudkar, S.V.; Gottesman, M.M.; Kimchi-Sarfaty, C. Silent (Synonymous) SNPs: Should We Care About Them? Methods Mol. Biol. 2009, 578, 23–39. [Google Scholar] [CrossRef]
- Lebeuf-Taylor, E.; McCloskey, N.; Bailey, S.; Hinz, A.; Kassen, R. The distribution of fitness effects among synonymous mutations in a gene under directional selection. eLife 2019, 8, e45952. [Google Scholar] [CrossRef]
- Chu, D.; Wei, L. Nonsynonymous, synonymous and nonsense mutations in human cancer-related genes undergo stronger purifying selections than expectation. BMC Cancer 2019, 19, 359. [Google Scholar] [CrossRef]
- Nei, M.; Maruyama, T.; Chakraborty, R. The Bottleneck Effect and Genetic Variability in Populations. Evolution 1975, 29, 1. [Google Scholar] [CrossRef]
- Liao, I. C.; Huang, Y. S. Methodological approach used for the domestication of potential candidates for aquaculture. Cah. Options Méditerranéennes 2000, 47, 97–107. [Google Scholar]
- Teletchea, F.; Fontaine, P. Levels of domestication in fish: Implications for the sustainable future of aquaculture. Fish Fish. 2012, 15, 181–195. [Google Scholar] [CrossRef]
- Dunham, R.A. Contribution of genetically improved aquatic organisms to global food security. In International Conference on Sustainable Contribution of Fisheries to Food Security, Sponsored by the Government of Japan and FAO; FAO: Rome, Italy, 1996; p. 150. [Google Scholar]
- Gutierrez, A.; Yáñez, J.; Davidson, W. Evidence of recent signatures of selection during domestication in an Atlantic salmon population. Mar. Genom. 2016, 26, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Cossu, P.; Scarpa, F.; Sanna, D.; Lai, T.; Dedola, G.L.; Curini-Galletti, M.; Mura, L.; Fois, N.; Casu, M. Influence of genetic drift on patterns of genetic variation: The footprint of aquaculture practices in Sparus aurata (Teleostei: Sparidae). Mol. Ecol. 2019, 28, 3012–3024. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Number of Reads | Raw | Trimmed | |
|---|---|---|---|
| Total | 345,567,818 | 323,864,488 | |
| Strains | Silver Steelhead | 170,172,570 | 159,237,809 |
| Born | 175,395,248 | 164,626,679 | |
| Tissue | Gills | 47,055,697 | 44,162,122 |
| Head kidney | 53,616,788 | 48,351,976 | |
| Heart | 52,003,354 | 49,537,095 | |
| Liver | 48,037,248 | 46,072,790 | |
| Muscle | 49,717,032 | 41,439,041 | |
| Spleen | 49,726,030 | 45,335,099 | |
| Strain | Categories | Exonic Region | ||||
|---|---|---|---|---|---|---|
| Exonic– Gene Symbol | Exonic– LOC Symbol | Others | CDS | 5-Prime Region | 3-Prime Region | |
| Silver Steelhead | 197 | 606 | 103 | 414 | 32 | 357 |
| Born | 64 | 224 | 35 | 160 | 15 | 113 |
| Total | 261 | 830 | 138 | 574 | 47 | 470 |
| Length of Protein-Coding Transcripts | Over All Tissues and Individuals |
|---|---|
| <1 kb | 62 |
| ≥1–<3 kb | 165 |
| ≥3–<5 kb | 23 |
| ≥5 kb | 11 |
| Total | 261 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
de los Ríos-Pérez, L.; Brunner, R.M.; Hadlich, F.; Rebl, A.; Kühn, C.; Wittenburg, D.; Goldammer, T.; Verleih, M. Comparative Analysis of the Transcriptome and Distribution of Putative SNPs in Two Rainbow Trout (Oncorhynchus mykiss) Breeding Strains by Using Next-Generation Sequencing. Genes 2020, 11, 841. https://doi.org/10.3390/genes11080841
de los Ríos-Pérez L, Brunner RM, Hadlich F, Rebl A, Kühn C, Wittenburg D, Goldammer T, Verleih M. Comparative Analysis of the Transcriptome and Distribution of Putative SNPs in Two Rainbow Trout (Oncorhynchus mykiss) Breeding Strains by Using Next-Generation Sequencing. Genes. 2020; 11(8):841. https://doi.org/10.3390/genes11080841
Chicago/Turabian Stylede los Ríos-Pérez, Lidia, Ronald Marco Brunner, Frieder Hadlich, Alexander Rebl, Carsten Kühn, Dörte Wittenburg, Tom Goldammer, and Marieke Verleih. 2020. "Comparative Analysis of the Transcriptome and Distribution of Putative SNPs in Two Rainbow Trout (Oncorhynchus mykiss) Breeding Strains by Using Next-Generation Sequencing" Genes 11, no. 8: 841. https://doi.org/10.3390/genes11080841