ShadowCaster: Compositional Methods under the Shadow of Phylogenetic Models to Detect Horizontal Gene Transfers in Prokaryotes


 , , , and
, , , and
Abstract
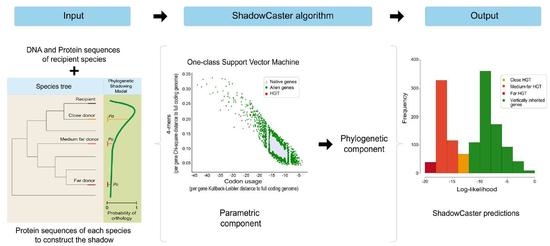
:
1. Introduction
2. Design and Implementation
2.1. Parametric Component
2.2. Phylogenetic Component
3. Results and Discussion
3.1. Performance on Simulated Data
- ShadowCaster identifies with the highest precision the HGT events from medium-far and far donors due to the differences in nucleotide composition content and orthologs. The recommended value of nu is 0.4.
- At the end of the parametric component, close HGT events are complex to classify within the group of atypical genes due to the similarity they share with the recipient sequence. This issue can be solved by incrementing the nu value but it is important to emphasize that will also increase the false positive rate (FPR).
- True and False Positive Rates did not change significantly with the increase of the number of proteomes/species in the phylogenetic shadow. The recommended starting number of proteomes is 15 since a small drop of the FPR was observed for the first time at this value. However, for extensive analyses in real datasets where the origin of the HGT is unknow, the number of proteomes could be increased up to 25. Considering that this parameter does not significantly affect the TPR and FPR, adding relevant information to the phylogenetic shadow tend to improve the quality of HGT detections. Please, see an example of ShadowCaster′s predictions belonging to its parametric and phylogenetic component on the real dataset used in the next section at https://github.com/dani2s/ShadowCaster_testData.
3.2. Performance on a Real Dataset. Comparison with the Most Popular State-Of-The-Art Computational Tools
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Andam, C.P.; Gogarten, J.P. Biased gene transfer in microbial evolution. Nat. Rev. Microbiol. 2011, 9, 543–555. [Google Scholar] [CrossRef] [PubMed]
- Hao, W.; Golding, G.B. The fate of laterally transferred genes: Life in the fast lane to adaptation or death. Genome Res. 2006, 16, 636–643. [Google Scholar] [CrossRef] [Green Version]
- Hemme, C.L.; Green, S.J.; Rishishwar, L.; Prakash, O.; Pettenato, A.; Chakraborty, R.; Deutschbauer, A.M.; Van Nostrand, J.D.; Wu, L.; He, Z.; et al. Lateral Gene Transfer in a Heavy Metal-Contaminated-Groundwater Microbial Community. MBio 2016, 7, e02234-15. [Google Scholar] [CrossRef] [Green Version]
- Ravenhall, M.; Škunca, N.; Lassalle, F.; Dessimoz, C. Inferring horizontal gene transfer. PLoS Comput. Biol. 2015, 11, e1004095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adato, O.; Ninyo, N.; Gophna, U.; Snir, S. Detecting Horizontal Gene Transfer between Closely Related Taxa. PLoS Comput. Biol. 2015, 11, e1004408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Becq, J.; Churlaud, C.; Deschavanne, P. A benchmark of parametric methods for horizontal transfers detection. PLoS ONE 2010, 5, e9989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poptsova, M.S.; Gogarten, J.P. The power of phylogenetic approaches to detect horizontally transferred genes. BMC Evol. Biol. 2007, 7, 45. [Google Scholar] [CrossRef] [Green Version]
- Zaneveld, J.R.; Nemergut, D.R.; Knight, R. Are all horizontal gene transfers created equal? Prospects for mechanism-based studies of HGT patterns. Microbiology 2008, 154, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Ragan, M.A.; Harlow, T.J.; Beiko, R.G. Do different surrogate methods detect lateral genetic transfer events of different relative ages? Trends Microbiol. 2006, 14, 4–8. [Google Scholar] [CrossRef]
- Koski, L.B.; Morton, R.A.; Golding, G.B. Codon bias and base composition are poor indicators of horizontally transferred genes. Mol. Biol. Evol. 2001, 18, 404–412. [Google Scholar] [CrossRef]
- Wang, B. Limitations of compositional approach to identifying horizontally transferred genes. J. Mol. Evol. 2001, 53, 244–250. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, M.; Ekstrom, A.; Li, X.; Yin, Y. HGT-Finder: A New Tool for Horizontal Gene Transfer Finding and Application to Aspergillus genomes. Toxins 2015, 7, 4035–4053. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Podell, S.; Gaasterland, T. DarkHorse: A method for genome-wide prediction of horizontal gene transfer. Genome Biol. 2007, 8, R16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Q.; Kosoy, M.; Dittmar, K. HGTector: An automated method facilitating genome-wide discovery of putative horizontal gene transfers. BMC Genom. 2014, 15, 717. [Google Scholar] [CrossRef] [Green Version]
- Ragan, M.A.; Beiko, R.G. Lateral genetic transfer: Open issues. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2009, 364, 2241–2251. [Google Scholar] [CrossRef]
- Vernikos, G.S.; Parkhill, J. Interpolated variable order motifs for identification of horizontally acquired DNA: Revisiting the Salmonella pathogenicity islands. Bioinformatics 2006, 22, 2196–2203. [Google Scholar] [CrossRef] [Green Version]
- Tsirigos, A.; Rigoutsos, I. A sensitive, support-vector-machine method for the detection of horizontal gene transfers in viral, archaeal and bacterial genomes. Nucleic Acids Res. 2005, 33, 3699–3707. [Google Scholar] [CrossRef]
- Ménigaud, S.; Mallet, L.; Picord, G.; Churlaud, C.; Borrel, A.; Deschavanne, P. GOHTAM: A website for “Genomic Origin of Horizontal Transfers, Alignment and Metagenomics”. Bioinformatics 2012, 28, 1270–1271. [Google Scholar]
- Xiong, D.; Xiao, F.; Liu, L.; Hu, K.; Tan, Y.; He, S.; Gao, X. Towards a better detection of horizontally transferred genes by combining unusual properties effectively. PLoS ONE 2012, 7, e43126. [Google Scholar] [CrossRef] [Green Version]
- Azad, R.K.; Lawrence, J.G. Towards more robust methods of alien gene detection. Nucleic Acids Res. 2011, 39, e56. [Google Scholar] [CrossRef] [Green Version]
- Schölkopf, B.; Platt, J.C.; Shawe-Taylor, J.; Smola, A.J.; Williamson, R.C. Estimating the support of a high-dimensional distribution. Neural Comput. 2001, 13, 1443–1471. [Google Scholar] [CrossRef]
- Gabaldón, T.; Koonin, E.V. Functional and evolutionary implications of gene orthology. Nat. Rev. Genet. 2013, 14, 360–366. [Google Scholar] [CrossRef] [Green Version]
- Li, L. OrthoMCL: Identification of Ortholog Groups for Eukaryotic Genomes. Genome Res. 2003, 13, 2178–2189. [Google Scholar] [CrossRef] [Green Version]
- Pál, C.; Papp, B.; Lercher, M.J. Adaptive evolution of bacterial metabolic networks by horizontal gene transfer. Nat. Genet. 2005, 37, 1372–1375. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Classification | Implementation | Methodological Highlights | Application Domain | Reference |
|---|---|---|---|---|
| Parametric methods | ||||
| Nucleotide composition | Alien Hunter (http://www.sanger.ac.uk/science/tools/alien-hunter) | Uses Interpolated Variable Order Motifs (IVOMs) coupled to a Hidden Markov Model (HMM) to detect alien (atypical genes). | bacterial genomes | [16] |
| No implementation available | Detects atypical genes based on k-mer (k = 8) frequencies using a one-class support vector machine (SVM). | viral, archaeal and bacterial genomes | [17] | |
| No implementation available | Combines two compositional features using a Kullback–Leibler divergence metric to improve the detection of atypical genes. | artificial genomes | [6] | |
| GOHTAM (http://gohtam.rpbs.univ-paris-diderot.fr/) | Uses a Jensen-Shannon divergence metric from window or gene-based signature data to detect atypical genes. | prokaryotic and eukaryotic genomes | [18] | |
| No implementation available | Detects atypical genes based on the selection of nine compositional features using a SVM. | bacterial genomes | [19] | |
| Nucleotide composition plus information from the genomic context | No implementation available | Implements a multiple-threshold approach to detect atypical genes from compositional features and genomic context information to reduce the chance of misclassification. | artificial genomes | [20] |
| Implicit phylogenetic methods | ||||
| Phyletic distributions based on BLAST searches | DarkHorse (http://darkhorse.ucsd.edu/) | Calculates a lineage probability index from BLAST searches to predict atypical genes. | prokaryotic and eukaryotic genomes. | [13] |
| HGTFinder (http://cys.bios.niu.edu/HGTFinder/HGTFinder.tar.gz) | Calculates a horizontal transfer index from BLAST searches to predict atypical genes. | prokaryotic and eukaryotic genomes. | [12] | |
| HGTector (https://github.com/DittmarLab/HGTector) | Establishes statistical thresholds to detect genes that do not adhere to a priori defined hierarchical evolutionary categories inferred from BLAST searches. | artificial, prokaryotic and eukaryotic genomes. | [14] | |
| Hybrid methods | ||||
| Nucleotide composition complemented with an implicit phylogenetic model | ShadowCaster | See further | prokaryotic genomes | This work |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sánchez-Soto, D.; Agüero-Chapin, G.; Armijos-Jaramillo, V.; Perez-Castillo, Y.; Tejera, E.; Antunes, A.; Sánchez-Rodríguez, A. ShadowCaster: Compositional Methods under the Shadow of Phylogenetic Models to Detect Horizontal Gene Transfers in Prokaryotes. Genes 2020, 11, 756. https://doi.org/10.3390/genes11070756
Sánchez-Soto D, Agüero-Chapin G, Armijos-Jaramillo V, Perez-Castillo Y, Tejera E, Antunes A, Sánchez-Rodríguez A. ShadowCaster: Compositional Methods under the Shadow of Phylogenetic Models to Detect Horizontal Gene Transfers in Prokaryotes. Genes. 2020; 11(7):756. https://doi.org/10.3390/genes11070756
Chicago/Turabian StyleSánchez-Soto, Daniela, Guillermin Agüero-Chapin, Vinicio Armijos-Jaramillo, Yunierkis Perez-Castillo, Eduardo Tejera, Agostinho Antunes, and Aminael Sánchez-Rodríguez. 2020. "ShadowCaster: Compositional Methods under the Shadow of Phylogenetic Models to Detect Horizontal Gene Transfers in Prokaryotes" Genes 11, no. 7: 756. https://doi.org/10.3390/genes11070756