The Importance of ATM and ATR in Physcomitrella patens DNA Damage Repair, Development, and Gene Targeting


Abstract
:1. Introduction
2. Materials and Methods
2.1. Identification and Characterisation of PpATM and PpATR Genes
2.2. Production of Ppatm and Ppatr Mutants
2.3. Plant Growth Assays
2.4. DNA Damage Resistance and Checkpoint Control Assays
2.5. Compilation of DNA Damage Repair-Related and ATM- and ATR-Interacting Genes from A. thaliana and P. patens
2.6. SuperSAGE and Transcriptomics
2.7. Gene Targeting Assays
2.8. Accession Numbers
3. Results
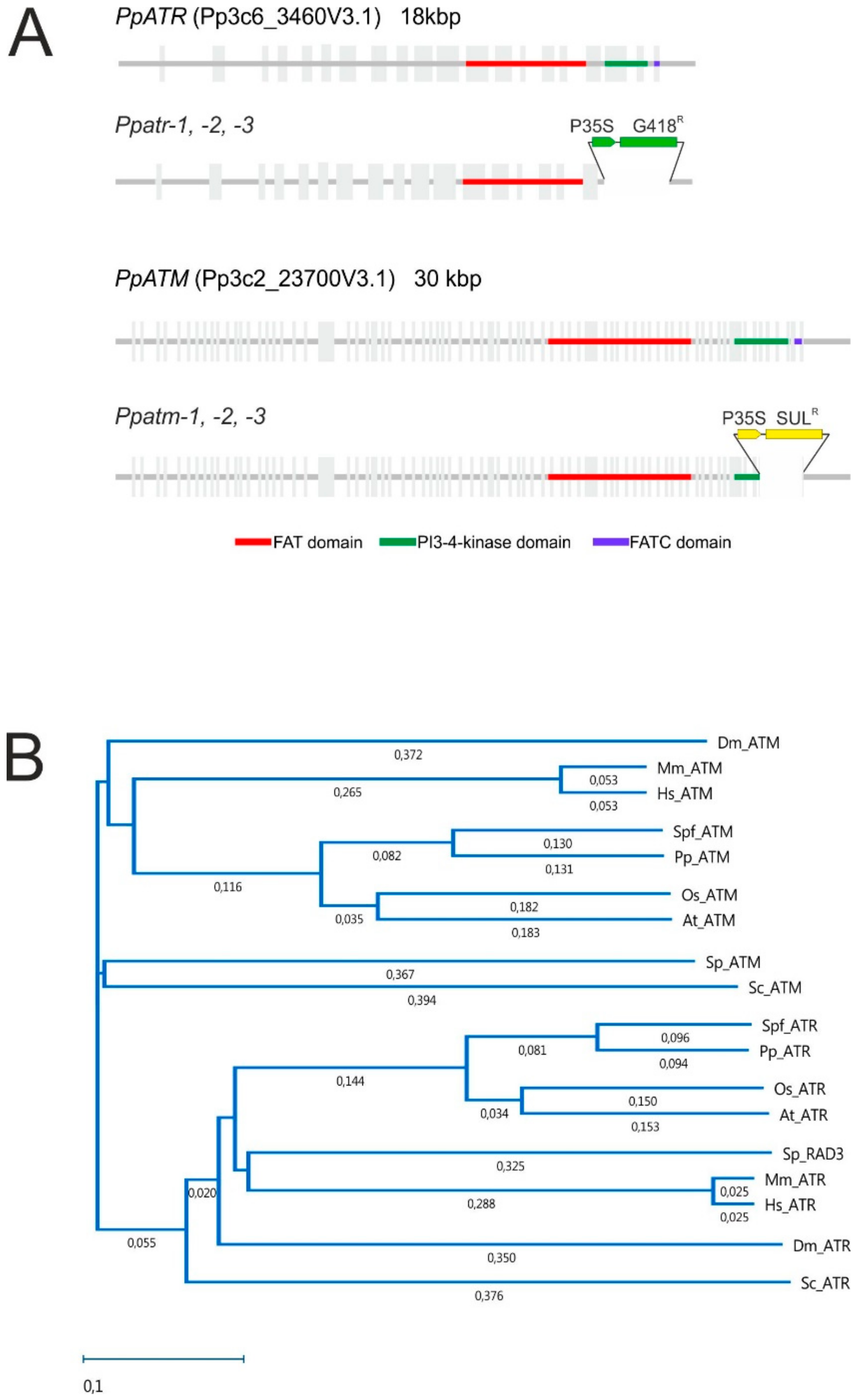
3.1. The P. patens ATM and ATR Orthologues, Gene Structure, and Production of Mutants
3.2. ATM and ATR are Important for Vegetative Development
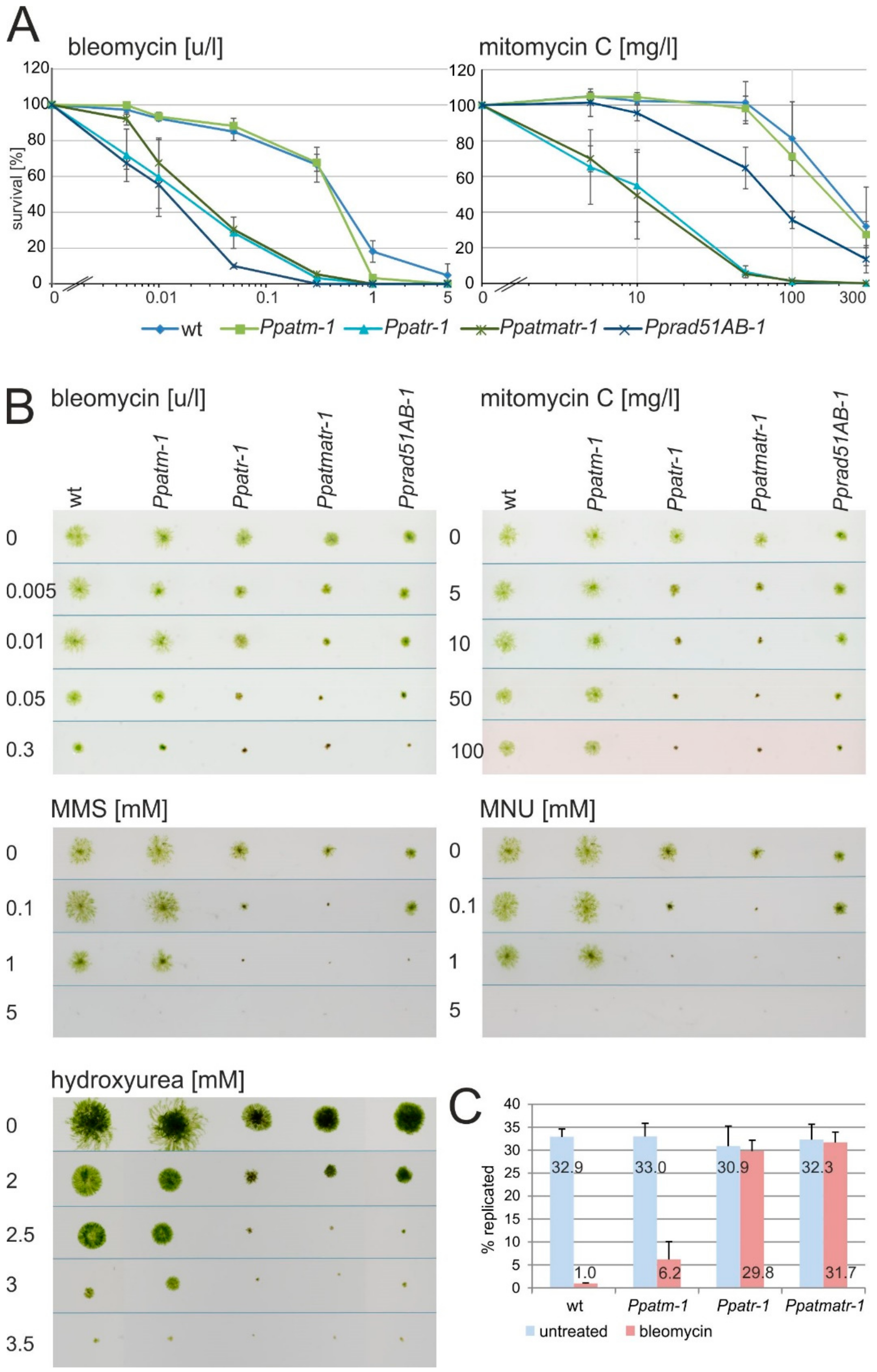
3.3. ATR Dominates DNA Damage Repair and Checkpoint Control
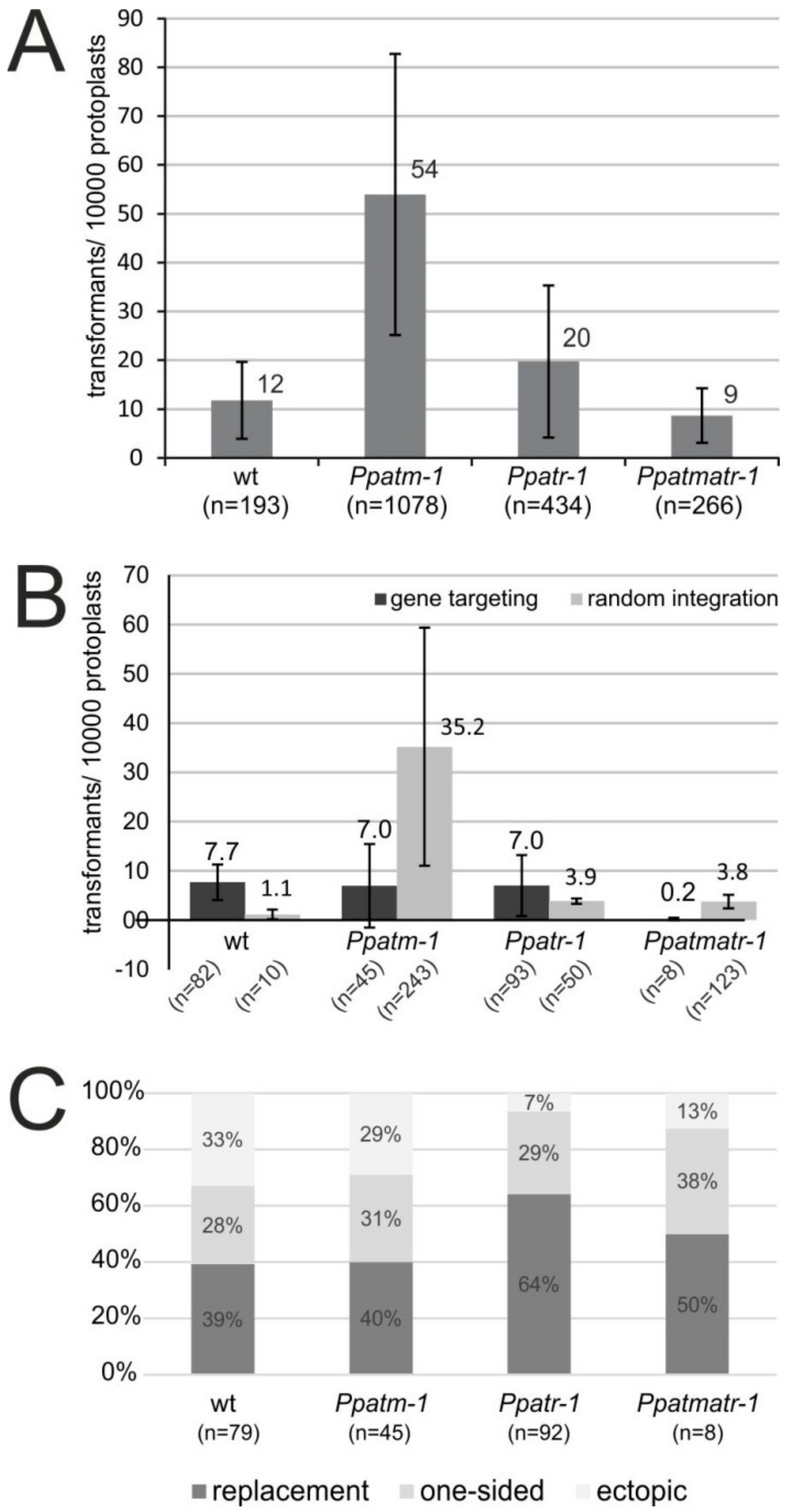
3.4. The Role of ATM and ATR in Gene Targeting
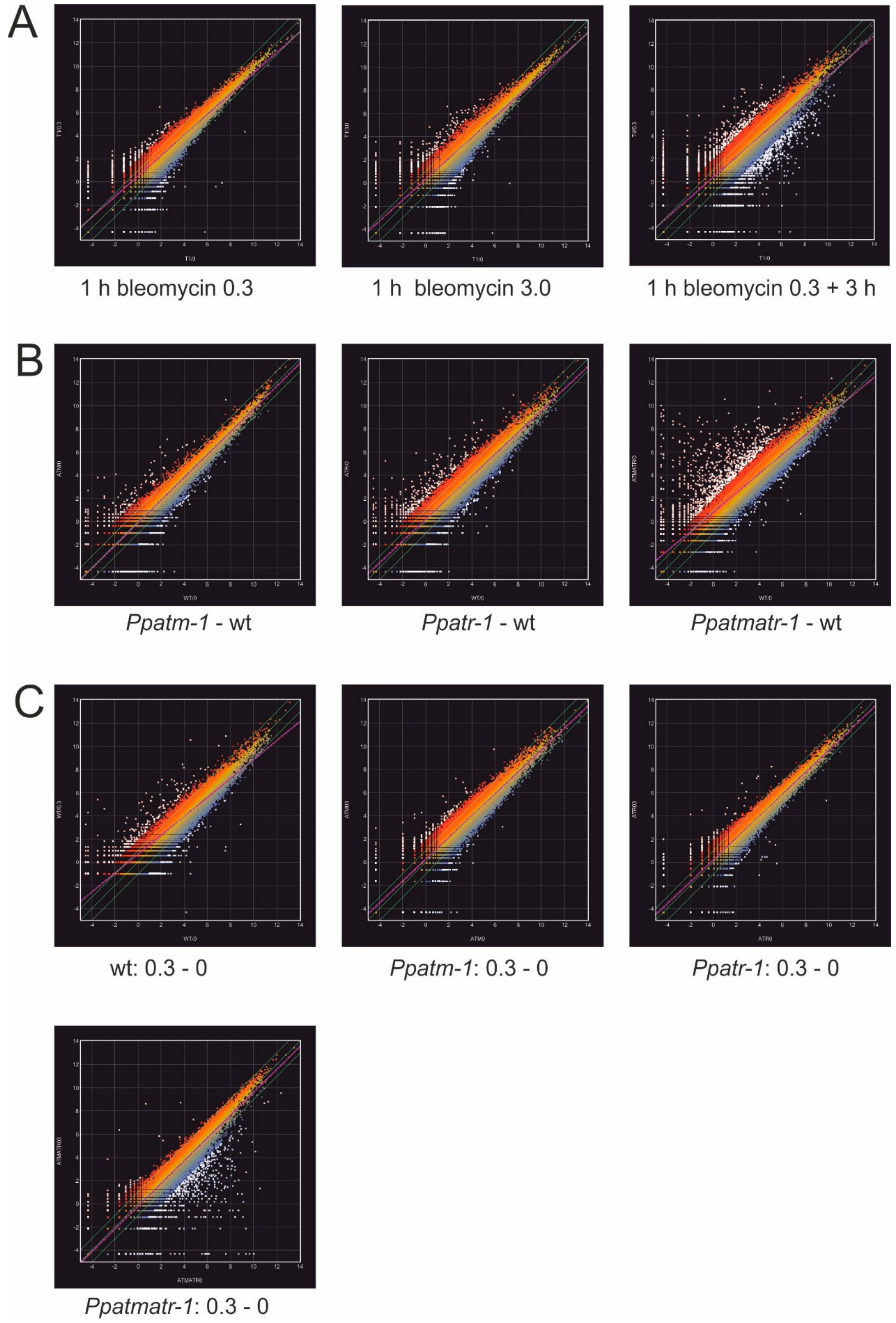
3.5. The Importance of ATM and ATR in the Transcriptional Response to Bleomycin-Induced Damage
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Tubbs, A.; Nussenzweig, A. Endogenous DNA Damage as a Source of Genomic Instability in Cancer. Cell 2017, 168, 644–656. [Google Scholar] [CrossRef]
- Melo, J.; Toczyski, D. A unified view of the DNA-damage checkpoint. Curr. Opin. Cell Biol. 2002, 14, 237–245. [Google Scholar] [CrossRef]
- Harper, J.W.; Elledge, S.J. The DNA Damage Response: Ten Years After. Mol. Cell 2007, 28, 739–745. [Google Scholar] [CrossRef]
- Ciccia, A.; Elledge, S.J. The DNA Damage Response: Making It Safe to Play with Knives. Mol. Cell 2010, 40, 179–204. [Google Scholar] [CrossRef] [Green Version]
- Hustedt, N.; Durocher, D. The control of DNA repair by the cell cycle. Nat. Cell Biol. 2017, 19, 1–9. [Google Scholar] [CrossRef]
- Heyer, W.D.; Ehmsen, K.T.; Liu, J. Regulation of homologous recombination in eukaryotes. Annu. Rev. Genet. 2010, 44, 113–139. [Google Scholar] [CrossRef] [Green Version]
- Ceccaldi, R.; Rondinelli, B.; D’Andrea, A.D. Repair Pathway Choices and Consequences at the Double-Strand Break. Trends Cell Biol. 2016, 26, 52–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shiloh, Y. ATM and related protein kinases: Safeguarding genome integrity. Nat. Rev. Cancer 2003, 3, 155–168. [Google Scholar] [CrossRef]
- Bensimon, A.; Aebersold, R.; Shiloh, Y. Beyond ATM: The protein kinase landscape of the DNA damage response. FEBS Lett. 2011, 585, 1625–1639. [Google Scholar] [CrossRef] [Green Version]
- Blackford, A.N.; Jackson, S.P. ATM, ATR, and DNA-PK: The Trinity at the Heart of the DNA Damage Response. Mol. Cell 2017, 66, 801–817. [Google Scholar] [CrossRef] [Green Version]
- Cimprich, K.A.; Cortez, D. ATR: An essential regulator of genome integrity. Nat. Rev. Mol. Cell Biol. 2008, 9, 616–627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saldivar, J.C.; Cortez, D.; Cimprich, K.A. The essential kinase ATR: Ensuring faithful duplication of a challenging genome. Nat. Rev. Mol. Cell Biol. 2017, 18, 622–636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia, V.; Bruchet, H.; Carnescasse, D.; Granier, F.; Bouchez, D.; Tissier, A. AtATM Is Essential for Meiosis and the Somatic Response to DNA Damage in Plants. Plant Cell 2002, 15, 119–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, C.H.; Finke, A.; Diaz, M.; Rozhon, W.; Poppenberger, B.; Baubec, T.; Pecinka, A. Repair of DNA Damage Induced by the Cytidine Analog Zebularine Requires ATR and ATM in Arabidopsis. Plant Cell 2015, 27, 1788–1800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Culligan, K.M.; Robertson, C.E.; Foreman, J.; Doerner, P.; Britt, A.B. ATR and ATM play both distinct and additive roles in response to ionizing radiation. Plant J. 2006, 48, 947–961. [Google Scholar] [CrossRef]
- Culligan, K.; Tissier, A.; Britt, A. ATR regulates a G2-phase cell-cycle checkpoint in Arabidopsis thaliana. Plant Cell 2004, 16, 1091–1104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, Z.; Cools, T.; De Veylder, L. Mechanisms Used by Plants to Cope with DNA Damage. Annu. Rev. Plant Biol. 2016, 67, 439–462. [Google Scholar] [CrossRef]
- Ricaud, L.; Proux, C.; Renou, J.-P.; Pichon, O.; Fochesato, S.; Ortet, P.; Montané, M.-H. ATM-Mediated Transcriptional and Developmental Responses to γ-rays in Arabidopsis. PLoS ONE 2007, 2, e430. [Google Scholar] [CrossRef]
- Cove, D. The Moss Physcomitrella patens. Annu. Rev. Genet. 2005, 39, 339–358. [Google Scholar] [CrossRef] [Green Version]
- Reski, R.; Cove, D.J. Physcomitrella patens. Curr. Biol. 2004, 14, R261–R262. [Google Scholar] [CrossRef] [Green Version]
- Rensing, S.A.; Goffinet, B.; Meyberg, R.; Wu, S.-Z.; Bezanilla, M. The Moss Physcomitrium (Physcomitrella) patens: A Model Organism for Non-Seed Plants. Plant Cell 2020, 32, 1361–1376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schaefer, D.G.; Zryd, J.P. Efficient gene targeting in the moss Physcomitrella patens. Plant J. 1997, 11, 1195–1206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goffova, I.; Vagnerova, R.; Peska, V.; Franek, M.; Havlova, K.; Hola, M.; Zachova, D.; Fojtova, M.; Cuming, A.; Kamisugi, Y.; et al. Roles of RAD51 and RTEL1 in telomere and rDNA stability in Physcomitrella patens. Plant J. 2019, 98, 1090–1105. [Google Scholar] [PubMed]
- Markmann-Mulisch, U.; Wendeler, E.; Zobell, O.; Schween, G.; Steinbiss, H.H.; Reiss, B. Differential requirements for RAD51 in Physcomitrella patens and Arabidopsis thaliana development and DNA damage repair. Plant Cell 2007, 19, 3080–3089. [Google Scholar] [CrossRef] [Green Version]
- Schaefer, D.G.; Delacote, F.; Charlot, F.; Vrielynck, N.; Guyon-Debast, A.; Le Guin, S.; Neuhaus, J.M.; Doutriaux, M.P.; Nogue, F. RAD51 loss of function abolishes gene targeting and de-represses illegitimate integration in the moss Physcomitrella patens. DNA Repair 2010, 9, 526–533. [Google Scholar] [CrossRef]
- Charlot, F.; Chelysheva, L.; Kamisugi, Y.; Vrielynck, N.; Guyon, A.; Epert, A.; Le Guin, S.; Schaefer, D.G.; Cuming, A.C.; Grelon, M.; et al. RAD51B plays an essential role during somatic and meiotic recombination in Physcomitrella. Nucleic Acids Res. 2014, 42, 11965–11978. [Google Scholar] [CrossRef] [Green Version]
- Kamisugi, Y.; Schaefer, D.G.; Kozak, J.; Charlot, F.; Vrielynck, N.; Hola, M.; Angelis, K.J.; Cuming, A.C.; Nogue, F. MRE11 and RAD50, but not NBS1, are essential for gene targeting in the moss Physcomitrella patens. Nucleic Acids Res. 2012, 40, 3496–3510. [Google Scholar] [CrossRef] [Green Version]
- Wiedemann, G.; van Gessel, N.; Kochl, F.; Hunn, L.; Schulze, K.; Maloukh, L.; Nogue, F.; Decker, E.L.; Hartung, F.; Reski, R. RecQ Helicases Function in Development, DNA Repair, and Gene Targeting in Physcomitrella patens. Plant Cell 2018, 30, 717–736. [Google Scholar] [CrossRef] [Green Version]
- Trouiller, B.; Schaefer, D.G.; Charlot, F.; Nogue, F. MSH2 is essential for the preservation of genome integrity and prevents homeologous recombination in the moss Physcomitrella patens. Nucleic Acids Res. 2006, 34, 232–242. [Google Scholar] [CrossRef]
- Wendeler, E.; Zobell, O.; Chrost, B.; Reiss, B. Recombination products suggest the frequent occurrence of aberrant gene replacement in the moss Physcomitrella patens. Plant J. 2015, 81, 548–558. [Google Scholar] [CrossRef]
- Collonnier, C.; Epert, A.; Mara, K.; Maclot, F.; Guyon-Debast, A.; Charlot, F.; White, C.; Schaefer, D.G.; Nogue, F. CRISPR-Cas9-mediated efficient directed mutagenesis and RAD51-dependent and RAD51-independent gene targeting in the moss Physcomitrella patens. Plant Biotech. J. 2017, 15, 122–131. [Google Scholar] [CrossRef] [PubMed]
- Rensing, S.A.; Lang, D.; Zimmer, A.D.; Terry, A.; Salamov, A.; Shapiro, H.; Nishiyama, T.; Perroud, P.-F.; Lindquist, E.A.; Kamisugi, Y.; et al. The Physcomitrella Genome Reveals Evolutionary Insights into the Conquest of Land by Plants. Science 2008, 319, 64–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lang, D.; Ullrich, K.K.; Murat, F.; Fuchs, J.; Jenkins, J.; Haas, F.B.; Piednoel, M.; Gundlach, H.; Van Bel, M.; Meyberg, R.; et al. The Physcomitrella patens chromosome-scale assembly reveals moss genome structure and evolution. Plant J. 2018, 93, 515–533. [Google Scholar] [CrossRef] [Green Version]
- Nordberg, H.; Cantor, M.; Dusheyko, S.; Hua, S.; Poliakov, A.; Shabalov, I.; Smirnova, T.; Grigoriev, I.V.; Dubchak, I. The genome portal of the Department of Energy Joint Genome Institute: 2014 updates. Nucleic Acids Res. 2014, 42, D26–D31. [Google Scholar] [CrossRef] [PubMed]
- Zobell, O.; Reiss, B. Gene targeting as a precise tool for plant mutagenesis. In The Handbook of Plant Mutation Screening (Mining of Natural and Induced Alleles); Meksem, K., Kahl, G., Eds.; Wiley-Blackwell-VCH Weinheim: Weinheim, Germany, 2010; pp. 263–285. [Google Scholar]
- Schween, G.; Gorr, G.; Hohe, A.; Reski, R. Unique tissue-specific cell cycle in Physcomitrella. Plant Biol. 2003, 5, 50–58. [Google Scholar] [CrossRef]
- Prakash, S.; Sung, P.; Prakash, L. DNA Repair Genes and Proteins of Saccharomyces Cerevisiae. Annu. Rev. Genet. 1993, 27, 33–70. [Google Scholar] [CrossRef]
- Wood, R.D.; Mitchell, M.; Lindahl, T. Human DNA repair genes, 2005. Mutat. Res./Fundam. Mol. Mech. Mutagenes 2005, 577, 275–283. [Google Scholar] [CrossRef]
- Markmann-Mulisch, U.; Hadi, M.Z.; Koepchen, K.; Alonso, J.C.; Russo, V.E.A.; Schell, J.; Reiss, B. The organization of Physcomitrella patens RAD51 genes is unique among eukaryotic organisms. Proc. Natl. Acad. Sci. USA 2002, 99, 2959–2964. [Google Scholar] [CrossRef] [Green Version]
- Barth, M.B.; Buchwalder, K.; Kawahara, A.Y.; Zhou, X.; Liu, S.; Krezdorn, N.; Rotter, B.; Horres, R.; Hundsdoerfer, A.K. Functional characterization of the Hyles euphorbiae hawkmoth transcriptome reveals strong expression of phorbol ester detoxification and seasonal cold hardiness genes. Front. Zool. 2018, 15, 20. [Google Scholar] [CrossRef] [Green Version]
- Stuckas, H.; Mende, M.B.; Hundsdoerfer, A.K. Response to cold acclimation in diapause pupae of Hyles euphorbiae (Lepidoptera: Sphingidae): Candidate biomarker identification using proteomics. Insect Mol. Biol. 2014, 23, 444–456. [Google Scholar] [CrossRef] [PubMed]
- Zimmer, A.D.; Lang, D.; Buchta, K.; Rombauts, S.; Nishiyama, T.; Hasebe, M.; Van de Peer, Y.; Rensing, S.A.; Reski, R. Reannotation and extended community resources for the genome of the non-seed plant Physcomitrella patens provide insights into the evolution of plant gene structures and functions. BMC Genom. 2013, 14, 498. [Google Scholar] [CrossRef] [Green Version]
- Cliby, W.A.; Roberts, C.J.; Cimprich, K.A.; Stringer, C.M.; Lamb, J.R.; Schreiber, S.L.; Friend, S.H. Overexpression of a kinase-inactive ATR protein causes sensitivity to DNA-damaging agents and defects in cell cycle checkpoints. EMBO J. 1998, 17, 159–169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daniel, J.A.; Pellegrini, M.; Lee, B.-S.; Guo, Z.; Filsuf, D.; Belkina, N.V.; You, Z.; Paull, T.T.; Sleckman, B.P.; Feigenbaum, L.; et al. Loss of ATM kinase activity leads to embryonic lethality in mice. J. Cell Biol. 2012, 198, 295–304. [Google Scholar] [CrossRef] [Green Version]
- Wright, J.A.; Keegan, K.S.; Herendeen, D.R.; Bentley, N.J.; Carr, A.M.; Hoekstra, M.F.; Concannon, P. Protein kinase mutants of human ATR increase sensitivity to UV and ionizing radiation and abrogate cell cycle checkpoint control. Proc. Natl. Acad. Sci. USA 1998, 95, 7445–7450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reski, R. Development, genetics and molecular biology of mosses. Bot. Acta 1998, 111, 1–15. [Google Scholar] [CrossRef]
- Moody, L.A.; Kelly, S.; Rabbinowitsch, E.; Langdale, J.A. Genetic Regulation of the 2D to 3D Growth Transition in the Moss Physcomitrella patens. Curr. Biol. 2018, 28, 473–478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kotogany, E.; Dudits, D.; Horvath, G.; Ayaydin, F. A rapid and robust assay for detection of S-phase cell cycle progression in plant cells and tissues by using ethynyl deoxyuridine. Plant Methods 2010, 6, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsumura, H.; Yoshida, K.; Luo, S.; Kimura, E.; Fujibe, T.; Albertyn, Z.; Barrero, R.A.; Krüger, D.H.; Kahl, G.; Schroth, G.P.; et al. High-Throughput SuperSAGE for Digital Gene Expression Analysis of Multiple Samples Using Next Generation Sequencing. PLoS ONE 2010, 5, e12010. [Google Scholar] [CrossRef] [Green Version]
- Molina, C.; Rotter, B.; Horres, R.; Udupa, S.M.; Besser, B.; Bellarmino, L.; Baum, M.; Matsumura, H.; Terauchi, R.; Kahl, G.; et al. SuperSAGE: The drought stress-responsive transcriptome of chickpea roots. BMC Genom. 2008, 9, 553. [Google Scholar] [CrossRef] [Green Version]
- Eichinger, C.S.; Jentsch, S. 9-1-1: PCNA’s specialized cousin. Trends Biochem. Sci. 2011, 36, 563–568. [Google Scholar] [CrossRef]
- Parrilla-Castellar, E.R.; Arlander, S.J.H.; Karnitz, L. Dial 9–1–1 for DNA damage: The Rad9–Hus1–Rad1 (9–1–1) clamp complex. DNA Repair 2004, 3, 1009–1014. [Google Scholar] [CrossRef] [PubMed]
- Huber, A.; Bai, P.; Murcia, J.M.D.; Murcia, G.D. PARP-1, PARP-2 and ATM in the DNA damage response: Functional synergy in mouse development. DNA Repair 2004, 3, 1103–1108. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Keppler, B.D.; Wise, R.R.; Bent, A.F. PARP2 Is the Predominant Poly(ADP-Ribose) Polymerase in Arabidopsis DNA Damage and Immune Responses. PLoS Genet. 2015, 11, e1005200. [Google Scholar] [CrossRef] [PubMed]
- Kamisugi, Y.; Whitaker, J.W.; Cuming, A.C. The Transcriptional Response to DNA-Double-Strand Breaks in Physcomitrella patens. PLoS ONE 2016, 11, e0161204. [Google Scholar] [CrossRef] [Green Version]
- Kass, E.M.; Helgadottir, H.R.; Chen, C.C.; Barbera, M.; Wang, R.; Westermark, U.K.; Ludwig, T.; Moynahan, M.E.; Jasin, M. Double-strand break repair by homologous recombination in primary mouse somatic cells requires BRCA1 but not the ATM kinase. Proc. Natl. Acad. Sci. USA 2013, 110, 5564–5569. [Google Scholar] [CrossRef] [Green Version]
- Mantiero, D.; Clerici, M.; Lucchini, G.; Longhese, M.P. Dual role for Saccharomyces cerevisiae Tel1 in the checkpoint response to double-strand breaks. EMBO Rep. 2007, 8, 380–387. [Google Scholar] [CrossRef] [Green Version]
- Craven, R.J.; Greenwell, P.W.; Dominska, M.; Petes, T.D. Regulation of Genome Stability by TEL1 and MEC1, Yeast Homologs of the Mammalian ATM and ATR Genes. Genetics 2002, 161, 493–507. [Google Scholar]
- Jaehnig, E.J.; Kuo, D.; Hombauer, H.; Ideker, T.G.; Kolodner, R.D. Checkpoint Kinases Regulate a Global Network of Transcription Factors in Response to DNA Damage. Cell Rep. 2013, 4, 174–188. [Google Scholar] [CrossRef] [Green Version]
- Sekelsky, J. DNA Repair in Drosophila: Mutagens, Models, and Missing Genes. Genetics 2017, 205, 471–490. [Google Scholar] [CrossRef] [Green Version]
- Ishikawa, M.; Murata, T.; Sato, Y.; Nishiyama, T.; Hiwatashi, Y.; Imai, A.; Kimura, M.; Sugimoto, N.; Akita, A.; Oguri, Y.; et al. Physcomitrella Cyclin-Dependent Kinase A Links Cell Cycle Reactivation to Other Cellular Changes during Reprogramming of Leaf Cells. Plant Cell 2011, 23, 2924–2938. [Google Scholar] [CrossRef] [Green Version]
- Liti, G. The fascinating and secret wild life of the budding yeast S. cerevisiae. eLife 2015, 4, e05835. [Google Scholar] [CrossRef] [PubMed]
- Bentley, N.J.; Holtzman, D.A.; Flaggs, G.; Keegan, K.S.; DeMaggio, A.; Ford, J.C.; Hoekstra, M.; Carr, A.M. The Schizosaccharomyces pombe rad3 checkpoint gene. EMBO J. 1996, 15, 6641–6651. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 1 | 2 | 3 | 4 | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Gene (UniProt nomenclature) | wt | Ppatm-1 | Ppatr-1 | Ppatmatr-1 | ||||||
| wt Bleo/wt0 | atm0/wt0 | atm Bleo/wt0 | atm Bleo/atm0 | atr0/wt0 | atr Bleo/wt0 | atr Bleo/atr0 | atmatr0/wt0 | atmatr Bleo/wt0 | atmatr Bleo/atmatr0 | |
| Checkpoints, Regulators, and Signaling | ||||||||||
| ATRIP | 0.8 | 0.9 | 0.5 | 0.6 | 0.8 | 0.8 | 1.0 | 1.0 | 0.9 | 0.8 |
| ATRX | 1.4 | 1.3 | 3.0 | 2.3 | 1.2 | 1.4 | 1.1 | 2.2 | 2.2 | 1.0 |
| hRAD1/REC1 | 0.6 | 0.6 | 0.4 | 0.7 | 0.4 | 0.6 | 1.6 | 0.4 | 0.4 | 0.9 |
| Hus1 | 2.6 | 0.7 | 3.8 | 5.1 | 1.8 | 1.2 | 0.7 | 2.0 | 1.8 | 0.9 |
| MAPK family | 1.5 | 0.9 | 0.6 | 0.7 | 1.0 | 1.2 | 1.1 | 1.2 | 1.6 | 1.3 |
| MDC1 | 2.6 | 0.6 | 1.9 | 3.0 | 2.0 | 1.6 | 0.8 | 3.8 | 2.1 | 0.6 |
| PARP2 | 6.0 | 0.8 | 2.6 | 3.5 | 1.0 | 1.2 | 1.2 | 3.0 | 0.7 | 0.3 |
| PMYT1 | 0.6 | 1.0 | 0.9 | 0.8 | 1.0 | 1.0 | 1.1 | 1.2 | 0.9 | 0.8 |
| RAD17 | 2.2 | 1.4 | 1.5 | 1.1 | 1.7 | 1.7 | 1.0 | 1.6 | 1.7 | 1.1 |
| RAD9A/Sprad9 | 21.0 | 0.4 | 10.4 | 27.6 | 2.6 | 2.4 | 0.9 | 4.7 | 2.3 | 0.5 |
| WEE1 | 0.9 | 0.7 | 0.9 | 1.4 | 1.1 | 1.7 | 1.5 | 0.7 | 1.1 | 1.7 |
| Homologous Recombination and Meiosis | ||||||||||
| HOP2 | 0.9 | 0.8 | 0.8 | 1.0 | 0.7 | 0.9 | 1.2 | 0.9 | 0.9 | 1.1 |
| MUS81 | 0.3 | 0.7 | 0.2 | 0.3 | 0.3 | 0.2 | 0.6 | 0.8 | 0.6 | 0.8 |
| NBN | 0.8 | 1.0 | 0.7 | 0.6 | 0.7 | 0.7 | 1.0 | 0.7 | 0.6 | 0.9 |
| PaxIP1 (1) | 0.8 | 0.8 | 0.7 | 0.8 | 1.0 | 0.8 | 0.9 | 1.8 | 0.8 | 0.4 |
| PaxIP1 (2) | 3.1 | 1.6 | 0.7 | 0.4 | 0.5 | 2.4 | 4.5 | 3.0 | 0.5 | 0.2 |
| PaxIP1 (3) | 0.5 | 0.7 | 0.7 | 1.0 | 0.9 | 0.6 | 0.6 | 1.1 | 1.0 | 0.9 |
| RA51B | 0.4 | 1.0 | 1.0 | 1.0 | 1.4 | 1.5 | 1.0 | 0.9 | 1.4 | 1.6 |
| RA51C | 1.8 | 0.7 | 0.8 | 1.0 | 1.3 | 1.6 | 1.2 | 1.2 | 1.3 | 1.1 |
| RA54B | 2.8 | 0.7 | 2.9 | 4.2 | 0.8 | 1.5 | 1.9 | 1.2 | 1.1 | 0.9 |
| RAD50 | 0.7 | 1.6 | 1.5 | 1.0 | 0.7 | 1.2 | 1.6 | 1.3 | 1.0 | 0.8 |
| PpRAD51A | 5.2 | 0.9 | 5.4 | 6.0 | 0.8 | 2.1 | 2.7 | 1.5 | 1.7 | 1.1 |
| PpRAD51B | 1.0 | 0.5 | 1.9 | 3.8 | 1.7 | 1.2 | 0.7 | 1.7 | 1.2 | 0.7 |
| RAD54 (1) | 2.8 | 0.7 | 3.5 | 4.9 | 1.4 | 1.6 | 1.2 | 2.1 | 2.2 | 1.0 |
| RAD54 (2) | 0.5 | 1.3 | 0.8 | 0.6 | 0.5 | 0.7 | 1.4 | 1.5 | 1.5 | 1.0 |
| RMI2 | 1.4 | 0.9 | 2.2 | 2.4 | 1.4 | 1.6 | 1.2 | 1.4 | 1.8 | 1.3 |
| RQSIM | 1.4 | 0.8 | 1.4 | 1.8 | 0.9 | 1.8 | 2.0 | 1.5 | 1.5 | 1.0 |
| SPO11 | 2.2 | 1.5 | 2.6 | 1.8 | 2.9 | 2.0 | 0.7 | 1.9 | 3.5 | 1.8 |
| TOP3B | 0.2 | 0.6 | 0.8 | 1.3 | 1.0 | 1.3 | 1.4 | 0.5 | 1.0 | 1.8 |
| WRIP1 (1) | 2.1 | 0.6 | 1.7 | 3.0 | 2.3 | 0.7 | 0.3 | 1.9 | 1.7 | 0.9 |
| WRIP1 (2) | 0.5 | 0.3 | 0.3 | 1.1 | 0.6 | 0.7 | 1.3 | 0.7 | 0.5 | 0.7 |
| WRX (1) | 1.3 | 1.1 | 1.5 | 1.4 | 3.3 | 2.1 | 0.6 | 3.0 | 1.7 | 0.6 |
| WRX (2) | 1.2 | 1.1 | 1.6 | 1.5 | 2.2 | 1.0 | 0.4 | 1.9 | 1.2 | 0.6 |
| XRCC2 | 0.7 | 0.6 | 0.9 | 1.6 | 0.7 | 0.9 | 1.2 | 1.6 | 1.5 | 1.0 |
| General Functions and Crosslink Repair | ||||||||||
| DNL1 | 1.5 | 1.0 | 0.9 | 0.8 | 0.6 | 0.8 | 1.4 | 0.8 | 0.8 | 0.9 |
| FANCL | 1.0 | 1.0 | 0.5 | 0.5 | 0.7 | 0.5 | 0.8 | 0.8 | 0.8 | 1.0 |
| PIF1 | 2.8 | 1.3 | 1.5 | 1.1 | 1.9 | 1.1 | 0.6 | 2.4 | 2.6 | 1.1 |
| PNKP | 4.6 | 0.5 | 5.5 | 10.6 | 0.4 | 1.3 | 3.3 | 2.7 | 2.9 | 1.1 |
| RAD21 | 0.5 | 0.7 | 1.1 | 1.5 | 0.5 | 0.8 | 1.5 | 0.7 | 0.5 | 0.7 |
| RNF4 (1) | 1.0 | 0.7 | 0.7 | 0.9 | 0.4 | 0.4 | 0.9 | 0.7 | 0.7 | 0.9 |
| RNF4 (2) | 1.1 | 1.4 | 1.5 | 1.1 | 1.3 | 1.1 | 0.8 | 2.9 | 1.6 | 0.5 |
| RNF4 (3) | 0.8 | 0.8 | 0.7 | 0.8 | 0.6 | 0.7 | 1.1 | 0.9 | 1.1 | 1.2 |
| RUVBL1 | 0.4 | 1.6 | 1.4 | 0.9 | 0.9 | 0.4 | 0.5 | 1.3 | 1.2 | 0.9 |
| RUVBL2 | 0.4 | 1.0 | 0.6 | 0.6 | 0.7 | 0.9 | 1.4 | 0.6 | 0.9 | 1.6 |
| SCC2 | 0.6 | 1.4 | 1.1 | 0.8 | 0.7 | 0.9 | 1.3 | 1.0 | 0.8 | 0.8 |
| SMC1A | 1.3 | 0.9 | 1.6 | 1.8 | 2.1 | 1.4 | 0.6 | 1.5 | 1.2 | 0.8 |
| SMC3 | 1.1 | 1.0 | 1.2 | 1.3 | 1.7 | 1.6 | 0.9 | 1.3 | 0.9 | 0.6 |
| SMC5 | 0.6 | 0.7 | 0.6 | 0.9 | 0.7 | 0.9 | 1.2 | 0.7 | 0.7 | 0.9 |
| SMC6 | 3.5 | 0.4 | 3.9 | 8.8 | 1.3 | 0.8 | 0.6 | 1.7 | 0.4 | 0.2 |
| STAG1 | 1.3 | 0.5 | 1.3 | 2.5 | 0.4 | 1.0 | 2.4 | 1.2 | 0.6 | 0.5 |
| Non-Homologous End-Joining | ||||||||||
| KU70/XRCC6 | 7.8 | 0.8 | 3.6 | 4.5 | 0.9 | 2.3 | 2.6 | 2.8 | 1.1 | 0.4 |
| KU80/XRCC5 | 5.3 | 0.9 | 4.3 | 4.8 | 2.5 | 2.3 | 0.9 | 3.8 | 3.3 | 0.9 |
| PRKDC | 0.9 | 1.3 | 1.3 | 1.0 | 0.9 | 0.9 | 1.1 | 0.5 | 0.4 | 0.8 |
| XRCC1 | 0.8 | 1.0 | 0.8 | 0.8 | 0.8 | 0.6 | 0.7 | 1.4 | 0.4 | 0.3 |
| XRCC4 | 5.1 | 0.2 | 2.5 | 11.1 | 0.3 | 1.2 | 4.8 | 0.4 | 0.7 | 1.6 |
| Mismatch Repair | ||||||||||
| EME1 | 0.6 | 1.3 | 1.6 | 1.2 | 2.3 | 3.0 | 1.3 | 1.4 | 3.0 | 2.1 |
| EXO1 | 1.2 | 1.0 | 1.3 | 1.4 | 1.4 | 1.6 | 1.2 | 1.3 | 1.5 | 1.2 |
| MLH1 | 1.7 | 0.8 | 1.2 | 1.4 | 1.0 | 1.2 | 1.1 | 1.4 | 0.9 | 0.7 |
| MLH3 | 2.5 | 0.8 | 1.0 | 1.2 | 0.8 | 1.2 | 1.6 | 1.4 | 1.0 | 0.7 |
| MSH2 | 1.3 | 0.4 | 1.5 | 3.9 | 1.4 | 1.3 | 1.0 | 1.8 | 1.5 | 0.9 |
| MSH3 | 1.4 | 1.0 | 1.2 | 1.2 | 1.3 | 0.9 | 0.7 | 1.5 | 0.7 | 0.5 |
| MSH5 | 0.9 | 0.9 | 1.1 | 1.3 | 1.1 | 1.5 | 1.4 | 1.3 | 1.3 | 1.0 |
| MSH6 | 0.4 | 0.4 | 0.9 | 2.2 | 0.4 | 1.1 | 2.5 | 0.9 | 0.7 | 0.8 |
| Muts | 0.6 | 1.5 | 1.5 | 1.0 | 1.5 | 1.3 | 0.9 | 2.2 | 2.3 | 1.1 |
| PMS1/PMS2 | 0.7 | 1.0 | 0.7 | 0.7 | 0.5 | 0.8 | 1.6 | 0.7 | 0.7 | 1.1 |
| Nucleotide Excision, Base Excision, and UV Repair | ||||||||||
| DDB1 (1) | 1.1 | 1.0 | 0.9 | 0.9 | 0.8 | 0.7 | 0.8 | 0.8 | 1.0 | 1.3 |
| DDB1 (2) | 0.8 | 1.0 | 1.5 | 1.5 | 1.4 | 1.2 | 0.9 | 1.1 | 1.1 | 1.0 |
| DDB2 | 3.6 | 0.9 | 2.5 | 2.8 | 1.6 | 1.5 | 0.9 | 2.9 | 1.0 | 0.3 |
| ERCC1/Rad10 (1) | 0.8 | 0.9 | 1.0 | 1.1 | 0.8 | 0.9 | 1.1 | 1.2 | 1.1 | 0.9 |
| ERCC1/Rad10 (2) | 1.1 | 0.3 | 0.4 | 1.3 | 1.3 | 0.2 | 0.2 | 5.0 | 0.3 | 0.1 |
| ERCC2/XPD | 2.4 | 0.5 | 1.8 | 3.6 | 1.1 | 0.9 | 0.8 | 1.6 | 1.8 | 1.1 |
| ERCC3/Rad25/XPB | 1.0 | 0.7 | 1.2 | 1.7 | 1.0 | 0.9 | 0.9 | 0.8 | 1.1 | 1.5 |
| ERCC5/RAD2 | 1.0 | 1.1 | 1.0 | 0.9 | 1.0 | 1.1 | 1.1 | 0.9 | 1.0 | 1.1 |
| ERCC6/rad26 (1) | 4.8 | 1.3 | 4.5 | 3.5 | 2.3 | 2.7 | 1.2 | 3.1 | 1.8 | 0.6 |
| ERCC6/rad26 (2) | 0.8 | 0.8 | 0.8 | 0.9 | 0.6 | 0.9 | 1.5 | 0.9 | 0.9 | 1.0 |
| RAD16 (1) | 1.3 | 0.7 | 2.2 | 3.1 | 1.8 | 1.8 | 1.0 | 2.2 | 1.9 | 0.9 |
| RAD16 (2) | 0.6 | 1.4 | 1.9 | 1.3 | 1.5 | 1.3 | 0.8 | 1.5 | 1.5 | 1.0 |
| RAD23 (1) | 0.5 | 0.9 | 0.7 | 0.7 | 0.7 | 0.8 | 1.3 | 0.8 | 0.8 | 1.0 |
| RAD23 (2) | 0.4 | 1.2 | 1.1 | 0.9 | 1.1 | 0.8 | 0.7 | 1.0 | 1.5 | 1.5 |
| RAD23 (3) | 1.9 | 1.1 | 1.2 | 1.1 | 1.6 | 0.7 | 0.4 | 2.4 | 1.1 | 0.5 |
| RAD27/FEN1 (1) | 3.5 | 1.3 | 1.7 | 1.3 | 0.9 | 2.0 | 2.3 | 2.0 | 2.8 | 1.4 |
| RAD27/FEN1 (2) | 2.1 | 0.8 | 1.4 | 1.7 | 0.4 | 1.5 | 4.3 | 1.4 | 1.0 | 0.7 |
| RAD6/UBC2 (1) | 1.0 | 1.1 | 1.1 | 1.1 | 1.1 | 1.1 | 1.0 | 1.5 | 1.1 | 0.7 |
| RAD6/UBC2 (2) | 1.0 | 0.6 | 0.7 | 1.1 | 0.8 | 0.9 | 1.1 | 0.8 | 0.8 | 1.0 |
| SYF1 | 1.5 | 1.1 | 0.8 | 0.7 | 1.1 | 1.4 | 1.3 | 1.3 | 1.6 | 1.2 |
| XPC/Rad4 | 1.1 | 0.3 | 2.9 | 10.0 | 1.5 | 1.6 | 1.1 | 0.2 | 0.8 | 4.2 |
| XPF/ERCC4/RAD1 | 1.5 | 0.5 | 1.4 | 3.0 | 1.2 | 1.1 | 1.0 | 1.1 | 1.4 | 1.3 |
| ATM and ATR Interactors | ||||||||||
| Cdc5l | 1.4 | 0.6 | 1.6 | 2.9 | 2.0 | 1.4 | 0.7 | 1.1 | 1.8 | 1.6 |
| CHD family (1) | 0.5 | 0.8 | 1.1 | 1.3 | 1.0 | 0.7 | 0.8 | 1.0 | 0.8 | 0.8 |
| CHD family (2) | 0.5 | 1.0 | 0.8 | 0.8 | 0.8 | 1.0 | 1.3 | 0.9 | 1.0 | 1.1 |
| E2F1 | 1.0 | 0.8 | 0.6 | 0.7 | 1.0 | 0.7 | 0.8 | 1.2 | 1.2 | 0.9 |
| H2A(1) | 1.1 | 1.4 | 2.0 | 1.4 | 1.4 | 1.1 | 0.8 | 1.5 | 1.4 | 0.9 |
| H2A (2) | 0.8 | 1.0 | 1.4 | 1.4 | 0.8 | 0.8 | 1.0 | 0.9 | 1.0 | 1.0 |
| HDAC1 (1) | 0.5 | 0.6 | 1.3 | 2.2 | 0.7 | 0.9 | 1.3 | 0.8 | 0.8 | 1.0 |
| HDAC1 (2) | 1.3 | 1.1 | 0.9 | 0.8 | 0.9 | 1.0 | 1.1 | 1.3 | 1.8 | 1.3 |
| HDAC1 (3) | 1.2 | 0.8 | 1.4 | 1.9 | 1.3 | 1.1 | 0.8 | 2.7 | 1.2 | 0.5 |
| MCM7 | 0.5 | 1.4 | 0.5 | 0.3 | 0.7 | 0.6 | 0.9 | 0.5 | 1.1 | 2.5 |
| PTPA (1) | 1.0 | 0.7 | 1.1 | 1.7 | 0.7 | 0.8 | 1.0 | 0.9 | 1.0 | 1.0 |
| PTPA (2) | 0.1 | 0.5 | 0.4 | 0.7 | 0.3 | 0.1 | 0.5 | 0.7 | 0.6 | 0.8 |
| RENT1 (UPF1) (1) | 0.4 | 0.6 | 1.0 | 1.7 | 0.8 | 1.5 | 1.9 | 0.6 | 1.5 | 2.6 |
| RENT1 (UPF1) (2) | 1.1 | 1.1 | 0.7 | 0.6 | 1.1 | 1.0 | 0.9 | 0.3 | 1.5 | 4.2 |
| RFA1 (RPA) | 1.2 | 0.8 | 2.3 | 2.8 | 0.8 | 1.1 | 1.4 | 1.1 | 0.8 | 0.8 |
| RFA2 (RPA) | 1.1 | 1.3 | 0.8 | 0.6 | 1.4 | 1.4 | 1.0 | 1.2 | 1.4 | 1.2 |
| SOSB1/hSSB1 | 0.6 | 1.2 | 1.1 | 0.9 | 0.8 | 1.4 | 1.6 | 0.6 | 0.8 | 1.3 |
| STRAP (1) | 0.6 | 1.1 | 0.4 | 0.4 | 1.3 | 0.7 | 0.6 | 0.9 | 0.7 | 0.8 |
| STRAP (2) | 0.7 | 0.7 | 0.7 | 0.9 | 0.8 | 0.8 | 1.1 | 0.9 | 0.7 | 0.8 |
| TERF family (1) | 0.6 | 0.7 | 0.4 | 0.6 | 0.5 | 0.6 | 1.2 | 0.5 | 0.6 | 1.2 |
| TERF family (2) | 1.8 | 1.1 | 1.9 | 1.7 | 1.9 | 1.4 | 0.7 | 1.9 | 0.8 | 0.4 |
| TERF family (3) | 0.8 | 0.9 | 0.6 | 0.6 | 1.0 | 0.9 | 0.9 | 1.0 | 1.1 | 1.1 |
| TERF family (4) | 2.2 | 0.8 | 1.4 | 1.7 | 1.9 | 2.5 | 1.3 | 2.1 | 2.2 | 1.1 |
| TERF family (5) | 1.9 | 0.7 | 0.9 | 1.3 | 1.0 | 1.0 | 1.0 | 1.3 | 0.3 | 0.2 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martens, M.; Horres, R.; Wendeler, E.; Reiss, B. The Importance of ATM and ATR in Physcomitrella patens DNA Damage Repair, Development, and Gene Targeting. Genes 2020, 11, 752. https://doi.org/10.3390/genes11070752
Martens M, Horres R, Wendeler E, Reiss B. The Importance of ATM and ATR in Physcomitrella patens DNA Damage Repair, Development, and Gene Targeting. Genes. 2020; 11(7):752. https://doi.org/10.3390/genes11070752
Chicago/Turabian StyleMartens, Martin, Ralf Horres, Edelgard Wendeler, and Bernd Reiss. 2020. "The Importance of ATM and ATR in Physcomitrella patens DNA Damage Repair, Development, and Gene Targeting" Genes 11, no. 7: 752. https://doi.org/10.3390/genes11070752