Comprehensive Geno- and Phenotyping in a Complex Pedigree Including Four Different Inherited Retinal Dystrophies

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
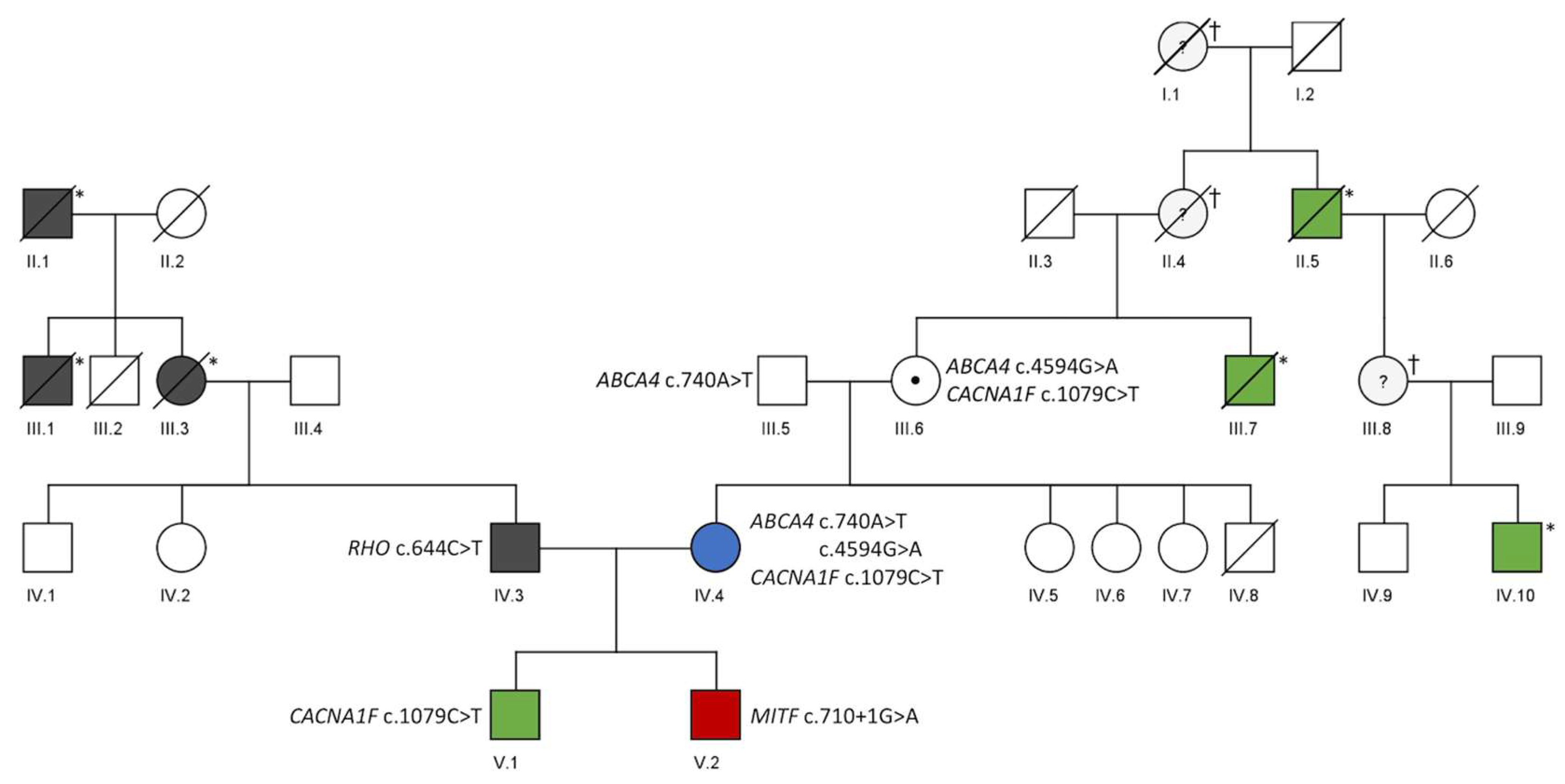
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Finger, R.P.; Fimmers, R.; Holz, F.G.; Scholl, H.P. Prevalence and causes of registered blindness in the largest federal state of Germany. Br. J. Ophthalmol. 2011, 95, 1061–1067. [Google Scholar] [CrossRef]
- Krumpaszky, H.G.; Ludtke, R.; Mickler, A.; Klauss, V.; Selbmann, H.K. Blindness incidence in Germany. A population-based study from Wurttemberg-Hohenzollern. Ophthalmologica 1999, 213, 176–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Birtel, J.; Gliem, M.; Holz, F.G.; Herrmann, P. Imaging and molecular genetic diagnostics for the characterization of retinal dystrophies. Ophthalmologe 2018, 115, 1021–1027. [Google Scholar] [CrossRef] [PubMed]
- Eisenberger, T.; Neuhaus, C.; Khan, A.O.; Decker, C.; Preising, M.N.; Friedburg, C.; Bieg, A.; Gliem, M.; Charbel Issa, P.; Holz, F.G.; et al. Increasing the yield in targeted next-generation sequencing by implicating CNV analysis, non-coding exons and the overall variant load: The example of retinal dystrophies. PLoS ONE 2013, 8, e78496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolz, H.J. Genetic diagnostics of retinal dystrophies: Breakthrough with new methods of DNA sequencing. Ophthalmologe 2018, 115, 1028–1034. [Google Scholar] [CrossRef]
- Shanks, M.E.; Downes, S.M.; Copley, R.R.; Lise, S.; Broxholme, J.; Hudspith, K.A.; Kwasniewska, A.; Davies, W.I.; Hankins, M.W.; Packham, E.R.; et al. Next-generation sequencing (NGS) as a diagnostic tool for retinal degeneration reveals a much higher detection rate in early-onset disease. Eur. J. Hum. Genet. 2013, 21, 274–280. [Google Scholar] [CrossRef] [Green Version]
- Glockle, N.; Kohl, S.; Mohr, J.; Scheurenbrand, T.; Sprecher, A.; Weisschuh, N.; Bernd, A.; Rudolph, G.; Schubach, M.; Poloschek, C.; et al. Panel-based next generation sequencing as a reliable and efficient technique to detect mutations in unselected patients with retinal dystrophies. Eur. J. Hum. Genet. 2014, 22, 99–104. [Google Scholar] [CrossRef]
- Oishi, M.; Oishi, A.; Gotoh, N.; Ogino, K.; Higasa, K.; Iida, K.; Makiyama, Y.; Morooka, S.; Matsuda, F.; Yoshimura, N. Next-generation sequencing-based comprehensive molecular analysis of 43 Japanese patients with cone and cone-rod dystrophies. Mol. Vis. 2016, 22, 150–160. [Google Scholar]
- Birtel, J.; Gliem, M.; Mangold, E.; Tebbe, L.; Spier, I.; Muller, P.L.; Holz, F.G.; Neuhaus, C.; Wolfrum, U.; Bolz, H.J.; et al. Novel Insights Into the Phenotypical Spectrum of KIF11-Associated Retinopathy, Including a New Form of Retinal Ciliopathy. Invest. Ophthalmol. Vis. Sci. 2017, 58, 3950–3959. [Google Scholar] [CrossRef] [Green Version]
- Bravo-Gil, N.; Gonzalez-Del Pozo, M.; Martin-Sanchez, M.; Mendez-Vidal, C.; Rodriguez-de la Rua, E.; Borrego, S.; Antinolo, G. Unravelling the genetic basis of simplex Retinitis Pigmentosa cases. Sci. Rep. 2017, 7, 41937. [Google Scholar] [CrossRef]
- Birtel, J.; Gliem, M.; Oishi, A.; Müller, P.L.; Herrmann, P.; Holz, F.G.; Mangold, E.; Knapp, M.; Bolz, H.J.; Charbel Issa, P. Genetic testing in patients with retinitis pigmentosa—Features of unsolved cases. Clin. Exp. Ophthalmol. 2019, 47, 779–786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Birtel, J.; Eisenberger, T.; Gliem, M.; Muller, P.L.; Herrmann, P.; Betz, C.; Zahnleiter, D.; Neuhaus, C.; Lenzner, S.; Holz, F.G.; et al. Clinical and genetic characteristics of 251 consecutive patients with macular and cone/cone-rod dystrophy. Sci. Rep. 2018, 8, 4824. [Google Scholar] [CrossRef] [PubMed]
- Birtel, J.; Gliem, M.; Mangold, E.; Muller, P.L.; Holz, F.G.; Neuhaus, C.; Lenzner, S.; Zahnleiter, D.; Betz, C.; Eisenberger, T.; et al. Next-generation sequencing identifies unexpected genotype-phenotype correlations in patients with retinitis pigmentosa. PLoS ONE 2018, 13, e0207958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yusuf, I.H.; Birtel, J.; Shanks, M.E.; Clouston, P.; Downes, S.M.; Charbel Issa, P.; MacLaren, R.E. Clinical Characterization of Retinitis Pigmentosa Associated with Variants in SNRNP200. JAMA Ophthalmol. 2019, in press. [Google Scholar] [CrossRef]
- De Castro-Miro, M.; Tonda, R.; Marfany, G.; Casaroli-Marano, R.P.; Gonzalez-Duarte, R. Novel mutation in the choroideremia gene and multi-Mendelian phenotypes in Spanish families. Br. J. Ophthalmol. 2018, 102, 1378–1386. [Google Scholar] [CrossRef]
- Cehajic-Kapetanovic, J.; Birtel, J.; McClements, M.E.; Shanks, M.E.; Clouston, P.; Downes, S.M.; Charbel Issa, P.; MacLaren, R.E. Clinical and Molecular Characterization of PROM1-Related Retinal Degeneration. JAMA Netw Open 2019, 2, e195752. [Google Scholar] [CrossRef] [Green Version]
- Preising, M.N.; Gorg, B.; Friedburg, C.; Qvartskhava, N.; Budde, B.S.; Bonus, M.; Toliat, M.R.; Pfleger, C.; Altmuller, J.; Herebian, D.; et al. Biallelic mutation of human SLC6A6 encoding the taurine transporter TAUT is linked to early retinal degeneration. FASEB J. 2019, 33, 11507–11527. [Google Scholar] [CrossRef]
- Martinez-Gimeno, M.; Trujillo, M.J.; Lorda, I.; Gimenez, A.; Calvo, M.T.; Ayuso, C.; Carballo, M. Three novel mutations (P215L, T289P, and 3811–2 A-->G) in the rhodopsin gene in autosomal dominant retinitis pigmentosa in Spanish families. Hum. Mutat. 2000, 16, 95–96. [Google Scholar] [CrossRef]
- Jaakson, K.; Zernant, J.; Kulm, M.; Hutchinson, A.; Tonisson, N.; Glavac, D.; Ravnik-Glavac, M.; Hawlina, M.; Meltzer, M.R.; Caruso, R.C.; et al. Genotyping microarray (gene chip) for the ABCR (ABCA4) gene. Hum. Mutat. 2003, 22, 395–403. [Google Scholar] [CrossRef]
- Lewis, R.A.; Shroyer, N.F.; Singh, N.; Allikmets, R.; Hutchinson, A.; Li, Y.; Lupski, J.R.; Leppert, M.; Dean, M. Genotype/Phenotype analysis of a photoreceptor-specific ATP-binding cassette transporter gene, ABCR, in Stargardt disease. Am. J. Hum. Genet. 1999, 64, 422–434. [Google Scholar] [CrossRef] [Green Version]
- Cella, W.; Greenstein, V.C.; Zernant-Rajang, J.; Smith, T.R.; Barile, G.; Allikmets, R.; Tsang, S.H. G1961E mutant allele in the Stargardt disease gene ABCA4 causes bull’s eye maculopathy. Exp. Eye Res. 2009, 89, 16–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pingault, V.; Ente, D.; Dastot-Le Moal, F.; Goossens, M.; Marlin, S.; Bondurand, N. Review and update of mutations causing Waardenburg syndrome. Hum. Mutat. 2010, 31, 391–406. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Li, X.; Shi, J.; Pang, X.; Hu, Y.; Wang, X.; Wu, H.; Yang, T. Molecular etiology and genotype-phenotype correlation of Chinese Han deaf patients with type I and type II Waardenburg Syndrome. Sci. Rep. 2016, 6, 35498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sullivan, L.S.; Bowne, S.J.; Birch, D.G.; Hughbanks-Wheaton, D.; Heckenlively, J.R.; Lewis, R.A.; Garcia, C.A.; Ruiz, R.S.; Blanton, S.H.; Northrup, H.; et al. Prevalence of disease-causing mutations in families with autosomal dominant retinitis pigmentosa: A screen of known genes in 200 families. Invest. Ophthalmol. Vis. Sci. 2006, 47, 3052–3064. [Google Scholar] [CrossRef] [PubMed]
- Daiger, S.P.; Bowne, S.J.; Sullivan, L.S. Genes and Mutations Causing Autosomal Dominant Retinitis Pigmentosa. Cold Spring Harb. Perspect. Med. 2014, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandez-San Jose, P.; Blanco-Kelly, F.; Corton, M.; Trujillo-Tiebas, M.J.; Gimenez, A.; Avila-Fernandez, A.; Garcia-Sandoval, B.; Lopez-Molina, M.I.; Hernan, I.; Carballo, M.; et al. Prevalence of Rhodopsin mutations in autosomal dominant Retinitis Pigmentosa in Spain: Clinical and analytical review in 200 families. Acta Ophthalmol. 2015, 93, e38–e44. [Google Scholar] [CrossRef]
- Audo, I.; Manes, G.; Mohand-Said, S.; Friedrich, A.; Lancelot, M.E.; Antonio, A.; Moskova-Doumanova, V.; Poch, O.; Zanlonghi, X.; Hamel, C.P.; et al. Spectrum of rhodopsin mutations in French autosomal dominant rod-cone dystrophy patients. Invest. Ophthalmol. Vis. Sci. 2010, 51, 3687–3700. [Google Scholar] [CrossRef]
- Heckenlively, J.R.; Rodriguez, J.A.; Daiger, S.P. Autosomal dominant sectoral retinitis pigmentosa. Two families with transversion mutation in codon 23 of rhodopsin. Arch. Ophthalmol. 1991, 109, 84–91. [Google Scholar] [CrossRef]
- Cideciyan, A.V.; Hood, D.C.; Huang, Y.; Banin, E.; Li, Z.Y.; Stone, E.M.; Milam, A.H.; Jacobson, S.G. Disease sequence from mutant rhodopsin allele to rod and cone photoreceptor degeneration in man. Proc. Natl. Acad. Sci. USA 1998, 95, 7103–7108. [Google Scholar] [CrossRef] [Green Version]
- Hamel, C.P. Cone rod dystrophies. Orphanet J. Rare Dis. 2007, 2, 7. [Google Scholar] [CrossRef] [Green Version]
- Kitiratschky, V.B.; Grau, T.; Bernd, A.; Zrenner, E.; Jagle, H.; Renner, A.B.; Kellner, U.; Rudolph, G.; Jacobson, S.G.; Cideciyan, A.V.; et al. ABCA4 gene analysis in patients with autosomal recessive cone and cone rod dystrophies. Eur. J. Hum. Genet. 2008, 16, 812–819. [Google Scholar] [CrossRef] [PubMed]
- Mata, N.L.; Weng, J.; Travis, G.H. Biosynthesis of a major lipofuscin fluorophore in mice and humans with ABCR-mediated retinal and macular degeneration. Proc. Natl. Acad. Sci. UA 2000, 97, 7154–7159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koenekoop, R.K. The gene for Stargardt disease, ABCA4, is a major retinal gene: A mini-review. Ophthalmic Genet. 2003, 24, 75–80. [Google Scholar] [CrossRef] [PubMed]
- . Cideciyan, A.V.; Aleman, T.S.; Swider, M.; Schwartz, S.B.; Steinberg, J.D.; Brucker, A.J.; Maguire, A.M.; Bennett, J.; Stone, E.M.; Jacobson, S.G. Mutations in ABCA4 result in accumulation of lipofuscin before slowing of the retinoid cycle: A reappraisal of the human disease sequence. Hum. Mol. Genet. 2004, 13, 525–534. [Google Scholar] [CrossRef]
- Duncker, T.; Tsang, S.H.; Lee, W.; Zernant, J.; Allikmets, R.; Delori, F.C.; Sparrow, J.R. Quantitative fundus autofluorescence distinguishes ABCA4-associated and non-ABCA4-associated bull’s-eye maculopathy. Ophthalmology 2015, 122, 345–355. [Google Scholar] [CrossRef] [Green Version]
- Bech-Hansen, N.T.; Naylor, M.J.; Maybaum, T.A.; Pearce, W.G.; Koop, B.; Fishman, G.A.; Mets, M.; Musarella, M.A.; Boycott, K.M. Loss-of-function mutations in a calcium-channel alpha1-subunit gene in Xp11.23 cause incomplete X-linked congenital stationary night blindness. Nat. Genet. 1998, 19, 264–267. [Google Scholar] [CrossRef]
- Strom, T.M.; Nyakatura, G.; Apfelstedt-Sylla, E.; Hellebrand, H.; Lorenz, B.; Weber, B.H.; Wutz, K.; Gutwillinger, N.; Ruther, K.; Drescher, B.; et al. An L-type calcium-channel gene mutated in incomplete X-linked congenital stationary night blindness. Nat. Genet. 1998, 19, 260–263. [Google Scholar] [CrossRef]
- Knoflach, D.; Schicker, K.; Glosmann, M.; Koschak, A. Gain-of-function nature of Cav1.4 L-type calcium channels alters firing properties of mouse retinal ganglion cells. Channels (Austin) 2015, 9, 298–306. [Google Scholar] [CrossRef] [Green Version]
- Hou, L.; Pavan, W.J. Transcriptional and signaling regulation in neural crest stem cell-derived melanocyte development: Do all roads lead to Mitf? Cell. Res. 2008, 18, 1163–1176. [Google Scholar] [CrossRef]
- Hughes, A.E.; Newton, V.E.; Liu, X.Z.; Read, A.P. A gene for Waardenburg syndrome type 2 maps close to the human homologue of the microphthalmia gene at chromosome 3p12-p14.1. Nat. Genet. 1994, 7, 509–512. [Google Scholar] [CrossRef]
- Tachibana, M.; Takeda, K.; Nobukuni, Y.; Urabe, K.; Long, J.E.; Meyers, K.A.; Aaronson, S.A.; Miki, T. Ectopic expression of MITF, a gene for Waardenburg syndrome type 2, converts fibroblasts to cells with melanocyte characteristics. Nat. Genet. 1996, 14, 50–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cortes-Gonzalez, V.; Zenteno, J.C.; Guzman-Sanchez, M.; Giordano-Herrera, V.; Guadarrama-Vallejo, D.; Ruiz-Quintero, N.; Villanueva-Mendoza, C. Tietz/Waardenburg type 2A syndrome associated with posterior microphthalmos in two unrelated patients with novel MITF gene mutations. Am. J. Med. Genet. A 2016, 170, 3294–3297. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.D.; Kelley, P.M.; Kenyon, J.B.; Hoover, D. Tietz syndrome (hypopigmentation/deafness) caused by mutation of MITF. J. Med. Genet. 2000, 37, 446–448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tietz, W. A syndrome of deaf-mutism associated with albinism showing dominant autosomal inheritance. Am. J. Hum. Genet. 1963, 15, 259–264. [Google Scholar] [PubMed]
- Bourneuf, E.; Otz, P.; Pausch, H.; Jagannathan, V.; Michot, P.; Grohs, C.; Piton, G.; Ammermuller, S.; Deloche, M.C.; Fritz, S.; et al. Rapid Discovery of De Novo Deleterious Mutations in Cattle Enhances the Value of Livestock as Model Species. Sci Rep. 2017, 7, 11466. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
| ID | Age | Gender | Refraction [dpt] (sph/cyl) | OD OS | BCVA | ODOS | Gene | Zygosity | Exons/ Introns | Nucleotide | Protein | Reference |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| IV.3 | 48 | m | −0.75/−2.25 −0.75/−1.00 | 20/32 20/50 | RHO | het | Exon 3 | c.644C>T | p.Pro215Leu | [18] | ||
| IV.4 | 45 | f | +3.00/−2.25 +3.25/−2.50 | HM HM | ABCA4 | het | Exon 6 | c.740A>T | p.(Asn247Ile) | [19] | ||
| het | Exon 31 | c.4594G>A | p.(Asp1532Asn) | [20,21] | ||||||||
| CACNA1F | het | Exon 8 | c.1079C>T | p.(Ser360Phe) | novel | |||||||
| V.1 | 13 | m | −1.75/−4.25 −2.75/−3.25 | 20/50 20/40 | CACNA1F | hem | Exon 8 | c.1079C>T | p.(Ser360Phe) | novel | ||
| V.2 | 11 | m | +4.50/−0.50 +6.00/−1.25 | 20/200 20/80 | MITF | het | Exon 7 | c.710+1G>A | splice site | [22] | ||
| ID | Gene | Nucleotide Change | Protein Change | MAF (GnomADEx.) | DANN | FATHMM | FATHMM-MKL | LRT | Mutation Assessor | Mutation Taster | PROVEAN | SIFT | HSF, SSPNN, varSEAK Online | Reference |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| IV.3 | RHO | c.644C>T | p.Pro215Leu | absent | 0.9989 | Tolerated | Damaging | Deleterious | High | Disease causing | Damaging | Damaging | [18] | |
| IV.4 | ABCA4 | c.740A>T | p.(Asn247Ile) | absent | 0.9905 | Damaging | Damaging | Neutral | Medium | Disease causing | Damaging | Damaging | - | [19] |
| c.4594G>A | p.(Asp1532Asn) | 0.000107 | 0.9993 | Damaging | Damaging | Deleterious | Medium | Disease causing | Damaging | Damaging | - | [20,21] | ||
| IV.4, V.1 | CACNA1F | c.1079C>T | p.(Ser360Phe) | absent | 0.9953 | Damaging | Damaging | Deleterious | Medium | Disease causing | Damaging | Damaging | - | novel |
| V.2 | MITF | c.710+1G>A | p.? | absent | 0.9956 | - | Damaging | - | - | Disease causing | - | - | LOF of donor splice site | [22] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Birtel, J.; Gliem, M.; Hess, K.; Birtel, T.H.; Holz, F.G.; Zechner, U.; Bolz, H.J.; Herrmann, P. Comprehensive Geno- and Phenotyping in a Complex Pedigree Including Four Different Inherited Retinal Dystrophies. Genes 2020, 11, 137. https://doi.org/10.3390/genes11020137
Birtel J, Gliem M, Hess K, Birtel TH, Holz FG, Zechner U, Bolz HJ, Herrmann P. Comprehensive Geno- and Phenotyping in a Complex Pedigree Including Four Different Inherited Retinal Dystrophies. Genes. 2020; 11(2):137. https://doi.org/10.3390/genes11020137
Chicago/Turabian StyleBirtel, Johannes, Martin Gliem, Kristina Hess, Theresa H. Birtel, Frank G. Holz, Ulrich Zechner, Hanno J. Bolz, and Philipp Herrmann. 2020. "Comprehensive Geno- and Phenotyping in a Complex Pedigree Including Four Different Inherited Retinal Dystrophies" Genes 11, no. 2: 137. https://doi.org/10.3390/genes11020137