GWAS Based on RNA-Seq SNPs and High-Throughput Phenotyping Combined with Climatic Data Highlights the Reservoir of Valuable Genetic Diversity in Regional Tomato Landraces

 ,
,  , , ,
, , ,  ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials and Experimental Design
2.2. Phenotypic Analysis
2.3. Carotenoids
2.4. Climatic Data
2.5. Transcriptome Sequencing and SNP Calling
2.6. Statistical and Population Genomic Analyses
2.7. Genome-Wide Association Studies
3. Results
3.1. Phenotypic Traits Analyses
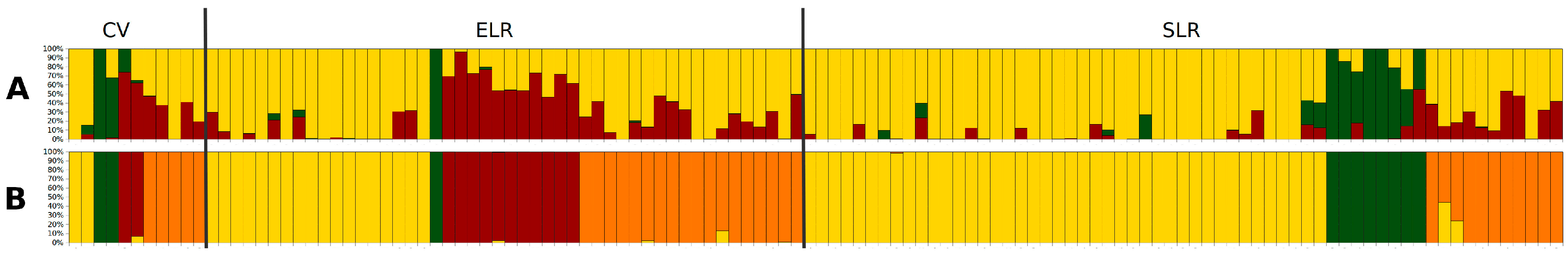
3.2. Genetic Diversity and Structure
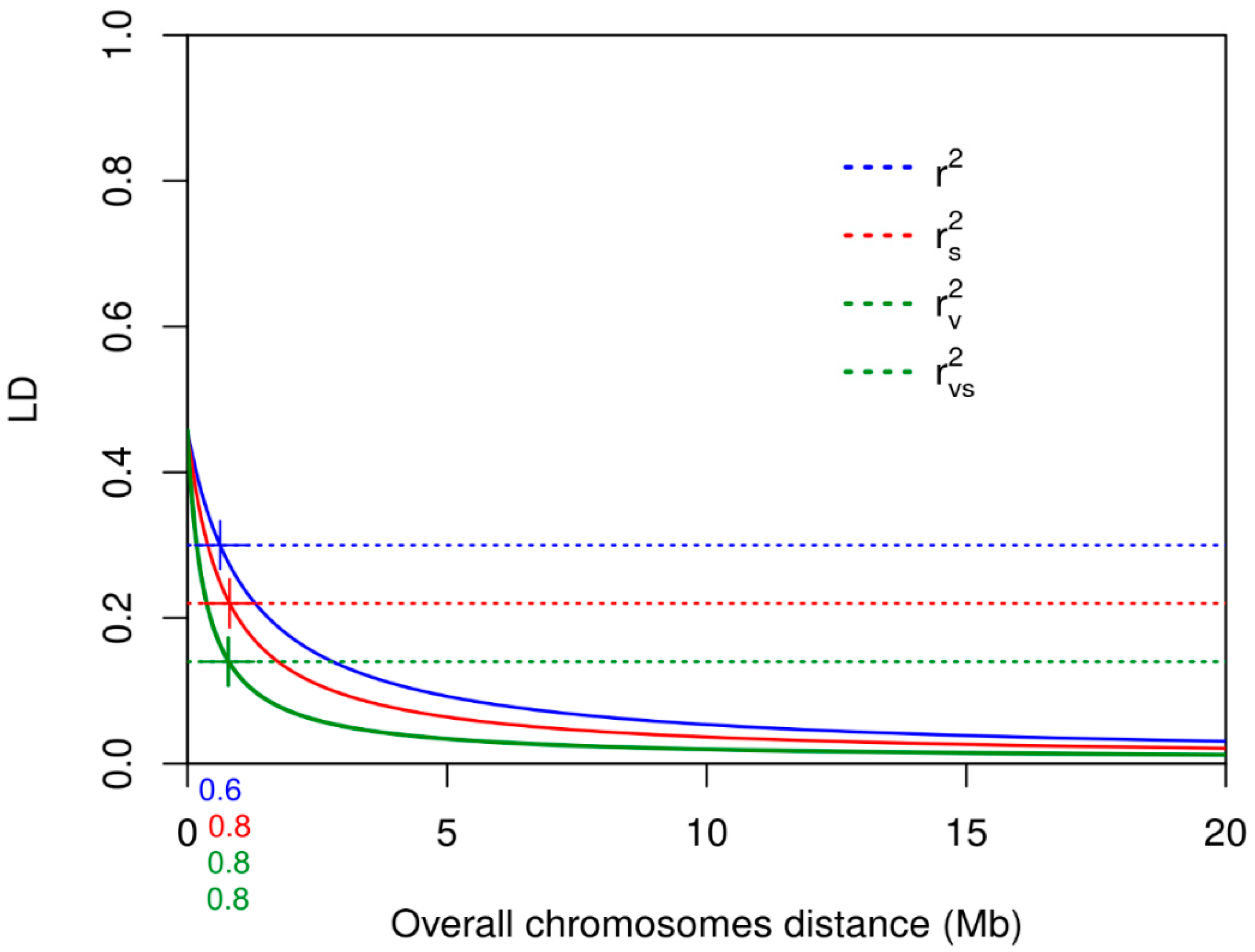
3.3. Linkage Disequilibrium
3.4. Genome-Wide Association Studies
3.5. Fruit and Plant Traits
3.6. Climatic Data
4. Discussion
4.1. Phenotypic and Molecular Diversity
4.2. Genome-Wide Association Study Results
4.3. Climatic Data
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Availability of Data and Material
References
- Gao, L.; Gonda, I.; Sun, H.; Ma, Q.; Bao, K.; Tieman, D.M.; Burzynski-Chang, E.A.; Fish, T.L.; Stromberg, K.A.; Sacks, G.L.; et al. The tomato pan-genome uncovers new genes and a rare allele regulating fruit flavor. Nat. Genet. 2019, 51, 1044–1051. [Google Scholar] [CrossRef]
- Lin, T.; Zhu, G.; Zhang, J.; Xu, X.; Yu, Q.; Zheng, Z.; Zhang, Z.; Lun, Y.; Li, S.; Wang, X.; et al. Genomic analyses provide insights into the history of tomato breeding. Nat. Genet. 2014, 46, 1220–1226. [Google Scholar] [CrossRef]
- Zhu, G.; Wang, S.; Huang, Z.; Zhang, S.; Liao, Q.; Zhang, C.; Lin, T.; Qin, M.; Peng, M.; Yang, C.; et al. Rewiring of the Fruit Metabolome in Tomato Breeding. Cell 2018, 172, 249–261.e12. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; Sauvage, C.; Zhao, J.; Bitton, F.; Bauchet, G.; Liu, D.; Huang, S.; Tieman, D.M.; Klee, H.J.; Causse, M. Meta-analysis of genome-wide association studies provides insights into genetic control of tomato flavor. Nat. Commun. 2019, 10, 1534. [Google Scholar] [CrossRef] [Green Version]
- Harlan, J.R. Our vanishing genetic resources. Science 1975, 188, 617–621. [Google Scholar]
- Brush, S.B. Genes in the Field: On-Farm Conservation of Crop Diversity; IDRC: Ottawa, ON, Canada, 2000. [Google Scholar]
- Petropoulos, S.A.; Barros, L.; Ferreira, I.C.F.R. Editorial: Rediscovering Local Landraces: Shaping Horticulture for the Future. Front. Plant Sci. 2019, 10, 126. [Google Scholar] [CrossRef] [Green Version]
- Hufford, M.B.; Berny Mier y Teran, J.C.; Gepts, P. Crop Biodiversity: An Unfinished Magnum Opus of Nature. Annu. Rev. Plant Biol. 2019, 70, 727–751. [Google Scholar] [CrossRef]
- McCouch, S. Diversifying selection in plant breeding. PLoS Biol. 2004, 2, e347. [Google Scholar]
- David, J.; Holtz, Y.; Ranwez, V.; Santoni, S.; Sarah, G.; Ardisson, M.; Poux, G.; Choulet, F.; Genthon, C.; Roumet, P.; et al. Genotyping by sequencing transcriptomes in an evolutionary pre-breeding durum wheat population. Mol. Breed. 2014, 34, 1531–1548. [Google Scholar] [CrossRef]
- Mazzucato, A.; Papa, R.; Bitocchi, E.; Mosconi, P.; Nanni, L.; Negri, V.; Picarella, M.E.; Siligato, F.; Soressi, G.P.; Tiranti, B.; et al. Genetic diversity, structure and marker-trait associations in a collection of Italian tomato (Solanum lycopersicum L.) landraces. Theor. Appl. Genet. 2008, 116, 657–669. [Google Scholar]
- Gupta, P.K.; Kulwal, P.L.; Jaiswal, V. Chapter Two—Association mapping in plants in the post-GWAS genomics era. In Advances in Genetics; Kumar, D., Ed.; Academic Press: Cambridge, MA, USA, 2019; Volume 104, pp. 75–154. [Google Scholar]
- Li, Z.; Wang, P.; You, C.; Yu, J.; Zhang, X.; Yan, F.; Ye, Z.; Shen, C.; Li, B.; Guo, K. Combined GWAS and eQTL analysis uncovers a genetic regulatory network orchestrating the initiation of secondary cell wall development in cotton. New Phytol. 2020, 226, 1738–1752. [Google Scholar]
- Sauvage, C.; Segura, V.; Bauchet, G.; Stevens, R.; Do, P.T.; Nikoloski, Z.; Fernie, A.R.; Causse, M. Genome-wide association in tomato reveals 44 candidate loci for fruit metabolic traits. Plant Physiol. 2014, 165, 1120–1132. [Google Scholar]
- Phan, N.T.; Trinh, L.T.; Rho, M.-Y.; Park, T.-S.; Kim, O.-R.; Zhao, J.; Kim, H.-M.; Sim, S.-C. Identification of loci associated with fruit traits using genome-wide single nucleotide polymorphisms in a core collection of tomato (Solanum lycopersicum L.). Sci. Hortic. 2019, 243, 567–574. [Google Scholar] [CrossRef]
- Sacco, A.; Ruggieri, V.; Parisi, M.; Festa, G.; Rigano, M.M.; Picarella, M.E.; Mazzucato, A.; Barone, A. Exploring a tomato landraces collection for fruit-related traits by the aid of a high-throughput genomic platform. PLoS ONE 2015, 10, e0137139. [Google Scholar]
- Yu, J.; Pressoir, G.; Briggs, W.H.; Bi, I.V.; Yamasaki, M.; Doebley, J.F.; McMullen, M.D.; Gaut, B.S.; Nielsen, D.M.; Holland, J.B.; et al. A unified mixed-model method for association mapping that accounts for multiple levels of relatedness. Nat. Genet. 2006, 38, 203–208. [Google Scholar] [CrossRef]
- Kang, H.M.; Zaitlen, N.A.; Wade, C.M.; Kirby, A.; Heckerman, D.; Daly, M.J.; Eskin, E. Efficient Control of Population Structure in Model Organism Association Mapping. Genetics 2008, 178, 1709–1723. [Google Scholar] [CrossRef] [Green Version]
- Segura, V.; Vilhjálmsson, B.J.; Platt, A.; Korte, A.; Seren, Ü.; Long, Q.; Nordborg, M. An efficient multi-locus mixed-model approach for genome-wide association studies in structured populations. Nat. Genet. 2012, 44, 825–830. [Google Scholar]
- Korte, A.; Farlow, A. The advantages and limitations of trait analysis with GWAS: A review. Plant Methods 2013, 9, 29. [Google Scholar]
- Liu, X.; Huang, M.; Fan, B.; Buckler, E.S.; Zhang, Z. Iterative usage of fixed and random effect models for powerful and efficient genome-wide association studies. PLoS Genet. 2016, 12, e1005767. [Google Scholar]
- Klasen, J.R.; Barbez, E.; Meier, L.; Meinshausen, N.; Bühlmann, P.; Koornneef, M.; Busch, W.; Schneeberger, K. A multi-marker association method for genome-wide association studies without the need for population structure correction. Nat. Commun. 2016, 7, 13299. [Google Scholar] [CrossRef] [Green Version]
- Louette, D. Traditional management of seed and genetic diversity: What is a landrace. Genes Field Fla. Lewis Publ. 2000, 109–142. [Google Scholar]
- Brewer, M.T.; Lang, L.; Fujimura, K.; Dujmovic, N.; Gray, S.; van der Knaap, E. Development of a controlled vocabulary and software application to analyze fruit shape variation in tomato and other plant species. Plant Physiol. 2006, 141, 15–25. [Google Scholar] [PubMed] [Green Version]
- Gonzalo, M.J.; Van Der Knaap, E. A comparative analysis into the genetic bases of morphology in tomato varieties exhibiting elongated fruit shape. Theor. Appl. Genet. 2008, 116, 647–656. [Google Scholar] [PubMed]
- Rodríguez, G.; Strecker, J.; Brewer, M.; Gonzalo, M.; Anderson, C.; Lang, L.; Sullivan, D.; Wagner, E.; Strecker, B.; Drushal, R.; et al. Tomato Analyzer Version 3 User Manual. 2010. Available online: https://vanderknaaplab.uga.edu/files/Tomato_Analyzer_3.0_Manual.pdf (accessed on 23 November 2020).
- Grassi, S.; Piro, G.; Lee, J.M.; Zheng, Y.; Fei, Z.; Dalessandro, G.; Giovannoni, J.J.; Lenucci, M.S. Comparative genomics reveals candidate carotenoid pathway regulators of ripening watermelon fruit. BMC Genom. 2013, 14, 781. [Google Scholar]
- Zhong, S.; Joung, J.-G.; Zheng, Y.; Chen, Y.; Liu, B.; Shao, Y.; Xiang, J.Z.; Fei, Z.; Giovannoni, J.J. High-Throughput Illumina Strand-Specific RNA Sequencing Library Preparation. Cold Spring Harb. Protoc. 2011, 2011, pdb.prot5652. [Google Scholar] [CrossRef] [Green Version]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinforma. Oxf. Engl. 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map format and SAMtools. Bioinforma. Oxf. Engl. 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Bates, D.M. lme4: Mixed-Effects Modeling with R; Springer: New York, NY, USA, 2010. [Google Scholar]
- Shannon, C.; Weaver, W. The Mathematical Theory of Communication; Urban University; Illinois Press: Champaign, IL, USA, 1949. [Google Scholar]
- Peakall, R.; Smouse, P.E. GENALEX 6: Genetic analysis in Excel. Population genetic software for teaching and research. Mol. Ecol. Notes 2006, 6, 288–295. [Google Scholar]
- Wei, T.; Simko, V. R Package “Corrplot”: Visualization of a Correlation Matrix (Version 0.84). Retrived Httpsgithub Comtaiyuncorrplot. 2017. Available online: https://github.com/taiyun/corrplot (accessed on 23 November 2020).
- Lawson, R.G.; Jurs, P.C. New index for clustering tendency and its application to chemical problems. J. Chem. Inf. Comput. Sci. 1990, 30, 36–41. [Google Scholar]
- Malika, C.; Ghazzali, N.; Boiteau, V.; Niknafs, A. NbClust: An R package for determining the relevant number of clusters in a data Set. J. Stat. Softw. 2014, 61, 1–36. [Google Scholar]
- Excoffier, L.; Lischer, H.E. Arlequin suite ver 3.5: A new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Resour. 2010, 10, 564–567. [Google Scholar]
- Jombart, T. Adegenet: A R package for the multivariate analysis of genetic markers. Bioinformatics 2008, 24, 1403–1405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pritchard, J.K.; Stephens, M.; Rosenberg, N.A.; Donnelly, P. Association mapping in structured populations. Am. J. Hum. Genet. 2000, 67, 170–181. [Google Scholar]
- Earl, D.A. STRUCTURE HARVESTER: A website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet. Resour. 2012, 4, 359–361. [Google Scholar]
- Privé, F.; Aschard, H.; Ziyatdinov, A.; Blum, M.G.B. Efficient analysis of large-scale genome-wide data with two R packages: Bigstatsr and bigsnpr. Bioinformatics 2018, 34, 2781–2787. [Google Scholar] [CrossRef] [Green Version]
- Bradbury, P.J.; Zhang, Z.; Kroon, D.E.; Casstevens, T.M.; Ramdoss, Y.; Buckler, E.S. TASSEL: Software for association mapping of complex traits in diverse samples. Bioinformatics 2007, 23, 2633–2635. [Google Scholar]
- Mangin, B.; Siberchicot, A.; Nicolas, S.; Doligez, A.; This, P.; Cierco-Ayrolles, C. Novel measures of linkage disequilibrium that correct the bias due to population structure and relatedness. Heredity 2012, 108, 285. [Google Scholar]
- Solé, X.; Guinó, E.; Valls, J.; Iniesta, R.; Moreno, V. SNPStats: A web tool for the analysis of association studies. Bioinformatics 2006, 22, 1928–1929. [Google Scholar]
- Clayton, D. SnpStats: SnpMatrix and XSnpMatrix Classes and Methods. R Package 2012. Available online: https://rdrr.io/bioc/snpStats/ (accessed on 23 November 2020).
- Marroni, F.; Pinosio, S.; Zaina, G.; Fogolari, F.; Felice, N.; Cattonaro, F.; Morgante, M. Nucleotide diversity and linkage disequilibrium in Populus nigra cinnamyl alcohol dehydrogenase (CAD4) gene. Tree Genet. Genomes 2011, 7, 1011–1023. [Google Scholar]
- Hill, W.G.; Weir, B.S. Variances and covariances of squared linkage disequilibria in finite populations. Theor. Popul. Biol. 1988, 33, 54–78. [Google Scholar] [PubMed]
- Barbato, M.; Orozco-terWengel, P.; Tapio, M.; Bruford, M.W. SNeP: A tool to estimate trends in recent effective population size trajectories using genome-wide SNP data. Front. Genet. 2015, 6, 109. [Google Scholar] [PubMed] [Green Version]
- Breseghello, F.; Sorrells, M.E. Association mapping of kernel size and milling quality in wheat (Triticum aestivum L.) cultivars. Genetics 2006, 172, 1165–1177. [Google Scholar] [PubMed] [Green Version]
- Lipka, A.E.; Tian, F.; Wang, Q.; Peiffer, J.; Li, M.; Bradbury, P.J.; Gore, M.A.; Buckler, E.S.; Zhang, Z. GAPIT: Genome association and prediction integrated tool. Bioinformatics 2012, 28, 2397–2399. [Google Scholar] [PubMed] [Green Version]
- Dahl, A.; Iotchkova, V.; Baud, A.; Johansson, Å.; Gyllensten, U.; Soranzo, N.; Mott, R.; Kranis, A.; Marchini, J. A multiple-phenotype imputation method for genetic studies. Nat. Genet. 2016, 48, 466–472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, S.; Zhang, B.; Keyhaninejad, N.; Rodríguez, G.R.; Kim, H.J.; Chakrabarti, M.; Illa-Berenguer, E.; Taitano, N.K.; Gonzalo, M.J.; Díaz, A.; et al. A common genetic mechanism underlies morphological diversity in fruits and other plant organs. Nat. Commun. 2018, 9, 4734. [Google Scholar] [CrossRef] [PubMed]
- Lippman, Z.; Tanksley, S.D. Dissecting the Genetic Pathway to Extreme Fruit Size in Tomato Using a Cross Between the Small-Fruited Wild Species Lycopersicon pimpinellifolium and L. esculentum var. Genetics 2001, 158, 413–422. [Google Scholar]
- Grandillo, S.; Ku, H.; Tanksley, S. Identifying the loci responsible for natural variation in fruit size and shape in tomato. Theor. Appl. Genet. 1999, 99, 978–987. [Google Scholar]
- Barrero, L.S.; Cong, B.; Wu, F.; Tanksley, S.D. Developmental characterization of the fasciated locus and mapping of Arabidopsis candidate genes involved in the control of floral meristem size and carpel number in tomato. Genome 2006, 49, 991–1006. [Google Scholar] [CrossRef]
- Cong, B.; Barrero, L.S.; Tanksley, S.D. Regulatory change in YABBY-like transcription factor led to evolution of extreme fruit size during tomato domestication. Nat. Genet. 2008, 40, 800–804. [Google Scholar] [CrossRef]
- Rodríguez, G.R.; Muños, S.; Anderson, C.; Sim, S.-C.; Michel, A.; Causse, M.; Gardener, B.B.M.; Francis, D.; van der Knaap, E. Distribution of SUN, OVATE, LC, and FAS in the tomato germplasm and the relationship to fruit shape diversity. Plant Physiol. 2011, 156, 275–285. [Google Scholar]
- Adato, A.; Mandel, T.; Mintz-Oron, S.; Venger, I.; Levy, D.; Yativ, M.; Domínguez, E.; Wang, Z.; De Vos, R.C.H.; Jetter, R.; et al. Fruit-Surface Flavonoid Accumulation in Tomato Is Controlled by a SlMYB12-Regulated Transcriptional Network. PLoS Genet. 2009, 5, e1000777. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Ye, H.; Guo, H.; Yin, Y. Arabidopsis IWS1 interacts with transcription factor BES1 and is involved in plant steroid hormone brassinosteroid regulated gene expression. Proc. Natl. Acad. Sci. USA 2010, 107, 3918–3923. [Google Scholar] [CrossRef] [Green Version]
- Reichheld, J.-P.; Khafif, M.; Riondet, C.; Droux, M.; Bonnard, G.; Meyer, Y. Inactivation of Thioredoxin Reductases Reveals a Complex Interplay between Thioredoxin and Glutathione Pathways in Arabidopsis Development. Plant Cell 2007, 19, 1851–1865. [Google Scholar] [CrossRef] [Green Version]
- Chi, Y.H.; Moon, J.C.; Park, J.H.; Kim, H.-S.; Zulfugarov, I.S.; Fanata, W.I.; Jang, H.H.; Lee, J.R.; Lee, Y.M.; Kim, S.T.; et al. Abnormal Chloroplast Development and Growth Inhibition in Rice Thioredoxin m Knock-Down Plants. Plant Physiol. 2008, 148, 808–817. [Google Scholar] [CrossRef] [Green Version]
- Meyer, Y.; Belin, C.; Delorme-Hinoux, V.; Reichheld, J.-P.; Riondet, C. Thioredoxin and glutaredoxin systems in plants: Molecular mechanisms, crosstalks, and functional significance. Antioxid. Redox Signal. 2012, 17, 1124–1160. [Google Scholar] [CrossRef]
- Bartoli, C.G.; Casalongué, C.A.; Simontacchi, M.; Marquez-Garcia, B.; Foyer, C.H. Interactions between hormone and redox signalling pathways in the control of growth and cross tolerance to stress. Environ. Exp. Bot. 2013, 94, 73–88. [Google Scholar] [CrossRef]
- Cheng, F.; Zhou, Y.-H.; Xia, X.-J.; Shi, K.; Zhou, J.; Yu, J.-Q. Chloroplastic thioredoxin-f and thioredoxin-m1/4 play important roles in brassinosteroids-induced changes in CO2 assimilation and cellular redox homeostasis in tomato. J. Exp. Bot. 2014, 65, 4335–4347. [Google Scholar] [CrossRef] [Green Version]
- Kumar, R.; Kushalappa, K.; Godt, D.; Pidkowich, M.S.; Pastorelli, S.; Hepworth, S.R.; Haughn, G.W. The Arabidopsis BEL1-LIKE HOMEODOMAIN Proteins SAW1 and SAW2 Act Redundantly to Regulate KNOX Expression Spatially in Leaf Margins. Plant Cell 2007, 19, 2719–2735. [Google Scholar] [CrossRef] [Green Version]
- Parnis, A.; Cohen, O.; Gutfinger, T.; Hareven, D.; Zamir, D.; Lifschitz, E. The dominant developmental mutants of tomato, Mouse-ear and Curl, are associated with distinct modes of abnormal transcriptional regulation of a Knotted gene. Plant Cell 1997, 9, 2143–2158. [Google Scholar] [CrossRef]
- Shani, E.; Burko, Y.; Ben-Yaakov, L.; Berger, Y.; Amsellem, Z.; Goldshmidt, A.; Sharon, E.; Ori, N. Stage-Specific Regulation of Solanum lycopersicum Leaf Maturation by Class 1 KNOTTED1-LIKE HOMEOBOX Proteins. Plant Cell 2009, 21, 3078–3092. [Google Scholar] [CrossRef] [Green Version]
- Mao, L.; Begum, D.; Chuang, H.; Budiman, M.A.; Szymkowiak, E.J.; Irish, E.E.; Wing, R.A. JOINTLESS is a MADS-box gene controlling tomato flower abscissionzone development. Nature 2000, 406, 910–913. [Google Scholar] [CrossRef]
- Mao, L.; Begum, D.; Goff, S.A.; Wing, R.A. Sequence and Analysis of the Tomato JOINTLESSLocus. Plant Physiol. 2001, 126, 1331–1340. [Google Scholar] [CrossRef] [Green Version]
- Roe, J.L.; Rivin, C.J.; Sessions, R.A.; Feldmann, K.A.; Zambryski, P.C. The Tousled gene in A. thaliana encodes a protein kinase homolog that is required for leaf and flower development. Cell 1993, 75, 939–950. [Google Scholar] [CrossRef]
- Uddin, M.N.; Dunoyer, P.; Schott, G.; Akhter, S.; Shi, C.; Lucas, W.J.; Voinnet, O.; Kim, J.-Y. The protein kinase TOUSLED facilitates RNAi in Arabidopsis. Nucleic Acids Res. 2014, 42, 7971–7980. [Google Scholar] [CrossRef] [Green Version]
- He, Y.; Doyle, M.R.; Amasino, R.M. PAF1-complex-mediated histone methylation of FLOWERING LOCUS C chromatin is required for the vernalization-responsive, winter-annual habit in Arabidopsis. Genes Dev. 2004, 18, 2774–2784. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.; Choi, K.; Park, C.; Hwang, H.-J.; Lee, I. SUPPRESSOR OF FRIGIDA4, Encoding a C2H2-Type Zinc Finger Protein, Represses Flowering by Transcriptional Activation of Arabidopsis FLOWERING LOCUS C. Plant Cell 2006, 18, 2985–2998. [Google Scholar] [CrossRef] [Green Version]
- Manning, K.; Tör, M.; Poole, M.; Hong, Y.; Thompson, A.J.; King, G.J.; Giovannoni, J.J.; Seymour, G.B. A naturally occurring epigenetic mutation in a gene encoding an SBP-box transcription factor inhibits tomato fruit ripening. Nat. Genet. 2006, 38, 948–952. [Google Scholar] [CrossRef]
- Preston, J.C.; Hileman, L. Functional Evolution in the Plant SQUAMOSA-PROMOTER BINDING PROTEIN-LIKE (SPL) Gene Family. Front. Plant Sci. 2013, 4, 80. [Google Scholar] [CrossRef] [Green Version]
- Silva, G.F.F.; Silva, E.M.; Correa, J.P.O.; Vicente, M.H.; Jiang, N.; Notini, M.M.; Junior, A.C.; Jesus, F.A.D.; Castilho, P.; Carrera, E.; et al. Tomato floral induction and flower development are orchestrated by the interplay between gibberellin and two unrelated microRNA-controlled modules. New Phytol. 2019, 221, 1328–1344. [Google Scholar] [CrossRef] [Green Version]
- Silva, G.F.F.; Silva, E.M.; Azevedo, M.D.S.; Guivin, M.A.C.; Ramiro, D.A.; Figueiredo, C.R.; Carrer, H.; Peres, L.E.P.; Nogueira, F.T.S. microRNA156-targeted SPL/SBP box transcription factors regulate tomato ovary and fruit development. Plant J. 2014, 78, 604–618. [Google Scholar] [CrossRef]
- Johanson, U.; West, J.; Lister, C.; Michaels, S.; Amasino, R.; Dean, C. Molecular Analysis of FRIGIDA, a Major Determinant of Natural Variation in Arabidopsis Flowering Time. Science 2000, 290, 344–347. [Google Scholar] [CrossRef]
- Gallavotti, A.; Long, J.A.; Stanfield, S.; Yang, X.; Jackson, D.; Vollbrecht, E.; Schmidt, R.J. The control of axillary meristem fate in the maize ramosa pathway. Dev. Camb. Engl. 2010, 137, 2849–2856. [Google Scholar] [CrossRef] [Green Version]
- Blanca, J.; Cañizares, J.; Cordero, L.; Pascual, L.; Diez, M.J.; Nuez, F. Variation revealed by SNP genotyping and morphology provides insight into the origin of the tomato. PLoS ONE 2012, 7, e48198. [Google Scholar]
- Sim, S.-C.; Van Deynze, A.; Stoffel, K.; Douches, D.S.; Zarka, D.; Ganal, M.W.; Chetelat, R.T.; Hutton, S.F.; Scott, J.W.; Gardner, R.G.; et al. High-Density SNP Genotyping of Tomato (Solanum lycopersicum L.) Reveals Patterns of Genetic Variation Due to Breeding. PLoS ONE 2012, 7, e45520. [Google Scholar] [CrossRef] [Green Version]
- Cortés-Olmos, C.; Vilanova, S.; Pascual, L.; Roselló, J.; Cebolla-Cornejo, J. SNP markers applied to the characterization of Spanish tomato (Solanum lycopersicum L.) landraces. Sci. Hortic. 2015, 194, 100–110. [Google Scholar] [CrossRef]
- Kalinowski, S.T. Counting alleles with rarefaction: Private alleles and hierarchical sampling designs. Conserv. Genet. 2004, 5, 539–543. [Google Scholar]
- Tanksley, S.D. The Genetic, Developmental, and Molecular Bases of Fruit Size and Shape Variation in Tomato. Plant Cell 2004, 16, S181–S189. [Google Scholar] [CrossRef]
- Colonna, V.; D’Agostino, N.; Garrison, E.; Albrechtsen, A.; Meisner, J.; Facchiano, A.; Cardi, T.; Tripodi, P. Genomic diversity and novel genome-wide association with fruit morphology in Capsicum, from 746k polymorphic sites. Sci. Rep. 2019, 9, 10067. [Google Scholar] [CrossRef] [Green Version]
- Bauchet, G.; Grenier, S.; Samson, N.; Segura, V.; Kende, A.; Beekwilder, J.; Cankar, K.; Gallois, J.-L.; Gricourt, J.; Bonnet, J. Identification of major loci and genomic regions controlling acid and volatile content in tomato fruit: Implications for flavor improvement. New Phytol. 2017, 215, 624–641. [Google Scholar]
- Ruggieri, V.; Francese, G.; Sacco, A.; D’Alessandro, A.; Rigano, M.M.; Parisi, M.; Milone, M.; Cardi, T.; Mennella, G.; Barone, A. An association mapping approach to identify favourable alleles for tomato fruit quality breeding. BMC Plant Biol. 2014, 14, 337. [Google Scholar]
- Cericola, F.; Portis, E.; Lanteri, S.; Toppino, L.; Barchi, L.; Acciarri, N.; Pulcini, L.; Sala, T.; Rotino, G.L. Linkage disequilibrium and genome-wide association analysis for anthocyanin pigmentation and fruit color in eggplant. BMC Genom. 2014, 15, 896. [Google Scholar] [CrossRef] [Green Version]
- Biselli, C.; Volante, A.; Desiderio, F.; Tondelli, A.; Gianinetti, A.; Finocchiaro, F.; Taddei, F.; Gazza, L.; Sgrulletta, D.; Cattivelli, L. GWAS for starch-related parameters in japonica rice (Oryza sativa L.). Plants 2019, 8, 292. [Google Scholar] [CrossRef] [Green Version]
- Garcia, M.; Eckermann, P.; Haefele, S.; Satija, S.; Sznajder, B.; Timmins, A.; Baumann, U.; Wolters, P.; Mather, D.E.; Fleury, D. Genome-wide association mapping of grain yield in a diverse collection of spring wheat (Triticum aestivum L.) evaluated in southern Australia. PLoS ONE 2019, 14, e0211730. [Google Scholar]
- Rogier, O.; Chateigner, A.; Amanzougarene, S.; Lesage-Descauses, M.-C.; Balzergue, S.; Brunaud, V.; Caius, J.; Soubigou-Taconnat, L.; Jorge, V.; Segura, V. Accuracy of RNAseq based SNP discovery and genotyping in Populus nigra. BMC Genom. 2018, 19, 909. [Google Scholar]
- Renaut, S.; Nolte, A.W.; Bernatchez, L. Mining transcriptome sequences towards identifying adaptive single nucleotide polymorphisms in lake whitefish species pairs (Coregonus spp. Salmonidae). Mol. Ecol. 2010, 19, 115–131. [Google Scholar] [PubMed]
- Pascual, L.; Albert, E.; Sauvage, C.; Duangjit, J.; Bouchet, J.-P.; Bitton, F.; Desplat, N.; Brunel, D.; Le Paslier, M.-C.; Ranc, N.; et al. Dissecting quantitative trait variation in the resequencing era: Complementarity of bi-parental, multi-parental and association panels. Plant Sci. 2016, 242, 120–130. [Google Scholar] [CrossRef] [Green Version]
- Pressoir, G.; Berthaud, J. Population structure and strong divergent selection shape phenotypic diversification in maize landraces. Heredity 2004, 92, 95–101. [Google Scholar] [CrossRef] [Green Version]
- Hamblin, M.T.; Casa, A.M.; Sun, H.; Murray, S.C.; Paterson, A.H.; Aquadro, C.F.; Kresovich, S. Challenges of Detecting Directional Selection After a Bottleneck: Lessons From Sorghum bicolor. Genetics 2006, 173, 953–964. [Google Scholar] [CrossRef] [Green Version]
- Labate, J.; Robertson, L.; Baldo, A. Multilocus sequence data reveal extensive departures from equilibrium in domesticated tomato (Solanum lycopersicum L.). Heredity 2009, 103, 257–267. [Google Scholar]
- Heerwaarden, J.V.; Eeuwijk, F.A.V.; Ross-Ibarra, J. Genetic diversity in a crop metapopulation. Heredity 2010, 104, 28–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Labate, J.A.; Sheffer, S.M.; Balch, T.; Robertson, L.D. Diversity and population structure in a geographic sample of tomato accessions. Crop Sci. 2011, 51, 1068–1079. [Google Scholar]
- Murgia, M.L.; Attene, G.; Rodriguez, M.; Bitocchi, E.; Bellucci, E.; Fois, D.; Nanni, L.; Gioia, T.; Albani, D.M.; Papa, R. A comprehensive phenotypic investigation of the “pod-shattering syndrome” in common bean. Front. Plant Sci. 2017, 8, 251. [Google Scholar] [PubMed] [Green Version]
- Xu, C.; Liberatore, K.L.; MacAlister, C.A.; Huang, Z.; Chu, Y.-H.; Jiang, K.; Brooks, C.; Ogawa-Ohnishi, M.; Xiong, G.; Pauly, M.; et al. A cascade of arabinosyltransferases controls shoot meristem size in tomato. Nat. Genet. 2015, 47, 784–792. [Google Scholar] [CrossRef]
- Muños, S.; Ranc, N.; Botton, E.; Bérard, A.; Rolland, S.; Duffé, P.; Carretero, Y.; Le Paslier, M.-C.; Delalande, C.; Bouzayen, M.; et al. Increase in Tomato Locule Number Is Controlled by Two Single-Nucleotide Polymorphisms Located Near WUSCHEL. Plant Physiol. 2011, 156, 2244–2254. [Google Scholar] [CrossRef] [Green Version]
- Huang, Z.; van der Knaap, E. Tomato fruit weight 11.3 maps close to fasciated on the bottom of chromosome 11. Theor. Appl. Genet. 2011, 123, 465–474. [Google Scholar] [CrossRef]
- Li, H.; Qi, M.; Sun, M.; Liu, Y.; Liu, Y.; Xu, T.; Li, Y.; Li, T. Tomato Transcription Factor SlWUS Plays an Important Role in Tomato Flower and Locule Development. Front. Plant Sci. 2017, 8. [Google Scholar] [CrossRef] [Green Version]
- Chu, Y.-H.; Jang, J.-C.; Huang, Z.; van der Knaap, E. Tomato locule number and fruit size controlled by natural alleles of lc and fas. Plant Direct 2019, 3, e00142. [Google Scholar] [CrossRef] [Green Version]
- Shirasawa, K.; Fukuoka, H.; Matsunaga, H.; Kobayashi, Y.; Kobayashi, I.; Hirakawa, H.; Isobe, S.; Tabata, S. Genome-Wide Association Studies Using Single Nucleotide Polymorphism Markers Developed by Re-Sequencing of the Genomes of Cultivated Tomato. DNA Res. 2013, 20, 593–603. [Google Scholar] [CrossRef] [Green Version]
- Chae, E.; Tan, Q.K.-G.; Hill, T.A.; Irish, V.F. An Arabidopsis F-box protein acts as a transcriptional co-factor to regulate floral development. Development 2008, 135, 1235–1245. [Google Scholar] [CrossRef] [Green Version]
- Bahrami, A.R.; Gray, J.E. Expression of a proteasome α-type subunit gene during tobacco development and senescence. Plant Mol. Biol. 1999, 39, 325–333. [Google Scholar] [PubMed]
- Astola, L.; Stigter, H.; van Dijk, A.D.J.; van Daelen, R.; Molenaar, J. Inferring the Gene Network Underlying the Branching of Tomato Inflorescence. PLoS ONE 2014, 9, e89689. [Google Scholar] [CrossRef]
- Ballester, A.-R.; Molthoff, J.; de Vos, R.; Hekkert, B.T.L.; Orzaez, D.; Fernández-Moreno, J.-P.; Tripodi, P.; Grandillo, S.; Martin, C.; Heldens, J.; et al. Biochemical and Molecular Analysis of Pink Tomatoes: Deregulated Expression of the Gene Encoding Transcription Factor SlMYB12 Leads to Pink Tomato Fruit Color. Plant Physiol. 2010, 152, 71–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, J.; Hu, T.; Yang, C.; Li, H.; Yang, M.; Ijaz, R.; Ye, Z.; Zhang, Y. Transcriptome Profiling of Tomato Fruit Development Reveals Transcription Factors Associated with Ascorbic Acid, Carotenoid and Flavonoid Biosynthesis. PLoS ONE 2015, 10, e0130885. [Google Scholar] [CrossRef]
- Gürbüz Çolak, N.; Eken, N.T.; Ülger, M.; Frary, A.; Doğanlar, S. Mapping of quantitative trait loci for antioxidant molecules in tomato fruit: Carotenoids, vitamins C and E, glutathione and phenolic acids. Plant Sci. 2020, 292, 110393. [Google Scholar] [CrossRef]
- Nakano, T.; Suzuki, K.; Fujimura, T.; Shinshi, H. Genome-wide analysis of the ERF gene family in Arabidopsis and rice. Plant Physiol. 2006, 140, 411–432. [Google Scholar] [CrossRef] [Green Version]
- Liu, M.; Gomes, B.L.; Mila, I.; Purgatto, E.; Peres, L.E.P.; Frasse, P.; Maza, E.; Zouine, M.; Roustan, J.-P.; Bouzayen, M.; et al. Comprehensive Profiling of Ethylene Response Factor Expression Identifies Ripening-Associated ERF Genes and Their Link to Key Regulators of Fruit Ripening in Tomato. Plant Physiol. 2016, 170, 1732–1744. [Google Scholar] [CrossRef] [Green Version]
- Phukan, U.J.; Jeena, G.S.; Tripathi, V.; Shukla, R.K. Regulation of Apetala2/Ethylene Response Factors in Plants. Front. Plant Sci. 2017, 8, 150. [Google Scholar] [CrossRef] [Green Version]
- Pujari, V.; Radebaugh, C.A.; Chodaparambil, J.V.; Muthurajan, U.M.; Almeida, A.R.; Fischbeck, J.A.; Luger, K.; Stargell, L.A. The transcription factor Spn1 regulates gene expression via a highly conserved novel structural motif. J. Mol. Biol. 2010, 404, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Petit, J.; Bres, C.; Mauxion, J.-P.; Tai, F.W.J.; Martin, L.B.B.; Fich, E.A.; Joubès, J.; Rose, J.K.C.; Domergue, F.; Rothan, C. The Glycerol-3-Phosphate Acyltransferase GPAT6 from Tomato Plays a Central Role in Fruit Cutin Biosynthesis. Plant Physiol. 2016, 171, 894–913. [Google Scholar] [CrossRef]
- Eshed, Y.; Baum, S.F.; Perea, J.V.; Bowman, J.L. Establishment of polarity in lateral organs of plants. Curr. Biol. 2001, 11, 1251–1260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khanday, I.; Yadav, S.R.; Vijayraghavan, U. Rice LHS1/OsMADS1 Controls Floret Meristem Specification by Coordinated Regulation of Transcription Factors and Hormone Signaling Pathways. Plant Physiol. 2013, 161, 1970–1983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, Y.; Zhang, K.; Guo, L.; Liu, X.; Zhang, Z. AUXIN RESPONSE FACTOR3 plays distinct role during early flower development. Plant Signal. Behav. 2018, 13, e1467690. [Google Scholar] [CrossRef] [Green Version]
- Hannon, E.; Weedon, M.; Bray, N.; O’Donovan, M.; Mill, J. Pleiotropic effects of trait-associated genetic variation on DNA methylation: Utility for refining GWAS loci. Am. J. Hum. Genet. 2017, 100, 954–959. [Google Scholar] [PubMed] [Green Version]
- Wang, W.; Feng, Z.; Bull, S.B.; Wang, Z. A 2-step strategy for detecting pleiotropic effects on multiple longitudinal traits. Front. Genet. 2014, 5, 357. [Google Scholar] [PubMed] [Green Version]
- Méndez-Vigo, B.; Picó, F.X.; Ramiro, M.; Martínez-Zapater, J.M.; Alonso-Blanco, C. Altitudinal and Climatic Adaptation Is Mediated by Flowering Traits and FRI, FLC, and PHYC Genes in Arabidopsis. Plant Physiol. 2011, 157, 1942–1955. [Google Scholar] [CrossRef] [Green Version]
- Facella, P.; Lopez, L.; Carbone, F.; Galbraith, D.W.; Giuliano, G.; Perrotta, G. Diurnal and Circadian Rhythms in the Tomato Transcriptome and Their Modulation by Cryptochrome Photoreceptors. PLoS ONE 2008, 3, e2798. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.-R.; Wang, T.-Y.; Weng, C.-P.; Duong, N.K.T.; Wu, S.-J. AtJ3, a specific HSP40 protein, mediates protein farnesylation-dependent response to heat stress in Arabidopsis. Planta 2019, 250, 1449–1460. [Google Scholar] [CrossRef]
- Li, Y.; Li, D.; Jiao, Y.; Schnable, J.C.; Li, Y.; Li, H.; Chen, H.; Hong, H.; Zhang, T.; Liu, B. Identification of loci controlling adaptation in Chinese soya bean landraces via a combination of conventional and bioclimatic GWAS. Plant Biotechnol. J. 2020, 18, 389–401. [Google Scholar]
- Sokolkova, A.; Bulyntsev, S.V.; Chang, P.L.; Carrasquilla-Garcia, N.; Igolkina, A.A.; Noujdina, N.V.; von Wettberg, E.; Vishnyakova, M.A.; Cook, D.R.; Nuzhdin, S.V. Genomic Analysis of Vavilov’s Historic Chickpea Landraces Reveals Footprints of Environmental and Human Selection. Int. J. Mol. Sci. 2020, 21, 3952. [Google Scholar] [CrossRef]
- Lasky, J.R.; Upadhyaya, H.D.; Ramu, P.; Deshpande, S.; Hash, C.T.; Bonnette, J.; Juenger, T.E.; Hyma, K.; Acharya, C.; Mitchell, S.E. Genome-environment associations in sorghum landraces predict adaptive traits. Sci. Adv. 2015, 1, e1400218. [Google Scholar] [PubMed] [Green Version]
- Du, L.; Poovaiah, B.W. A Novel Family of Ca 2+ /Calmodulin-Binding Proteins Involved in Transcriptional Regulation: Interaction with fsh/Ring3 Class Transcription Activators. Plant Mol. Biol. 2004, 54, 549–569. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.; Henderson, D.A.; Zhu, J.-K. The Arabidopsis Cold-Responsive Transcriptome and Its Regulation by ICE1. Plant Cell 2005, 17, 3155–3175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faye, J.M.; Maina, F.; Hu, Z.; Fonceka, D.; Cisse, N.; Morris, G.P. Genomic signatures of adaptation to Sahelian and Soudanian climates in sorghum landraces of Senegal. Ecol. Evol. 2019, 9, 6038–6051. [Google Scholar] [PubMed]
- Min, D.; Li, F.; Zhang, X.; Cui, X.; Shu, P.; Dong, L.; Ren, C. SlMYC2 Involved in Methyl Jasmonate-Induced Tomato Fruit Chilling Tolerance. J. Agric. Food Chem. 2018, 66, 3110–3117. [Google Scholar] [CrossRef]
- Giovannoni, J. Tomato Multiomics Reveals Consequences of Crop Domestication and Improvement. Cell 2018, 172, 6–8. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Trait | Year | Genotype | Genotype × Year | h2B | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| DF | SS | F | DF | SS | F | DF | SS | F | 2012 | 2013 | 2012/2013 | ||||
| DTFs | 1 | 508,837.7 | 26,547.5 | **** | 121 | 43,086.7 | 18.6 | **** | 121 | 15,408.1 | 6.6 | **** | 51.2 | 55.0 | 34.4 |
| DTFt | 1 | 125,556.0 | 6876.4 | **** | 121 | 41,182.4 | 18.6 | **** | 121 | 14,045.8 | 6.4 | **** | 51.2 | 58.0 | 35.9 |
| FRI | 1 | 247,271.0 | 7000.0 | **** | 121 | 31,918.6 | 7.5 | **** | 121 | 22,707.8 | 5.3 | **** | 30.7 | 43.4 | 11.8 |
| NFI | 1 | 170.8 | 8.3 | ** | 121 | 25,164.3 | 10.1 | **** | 121 | 8662.5 | 3.5 | **** | 65.7 | 57.6 | 36.3 |
| LLE | 1 | 577.3 | 36.5 | **** | 121 | 12,356.6 | 6.5 | **** | 121 | 3732.4 | 1.9 | **** | 42.5 | 25.9 | 28.6 |
| LWI | 1 | 2.2 | 0.1 | n.s. | 121 | 19,444.8 | 6.8 | **** | 121 | 6552.4 | 2.3 | **** | 43.9 | 31.7 | 27.3 |
| LL/W | 1 | 0.6 | 29.7 | **** | 121 | 10.0 | 4.1 | **** | 121 | 3.9 | 1.6 | **** | 26.0 | 42.1 | 20.0 |
| FWG | 1 | 403.6 | 0.1 | n.s. | 121 | 13,339,038.6 | 33.2 | **** | 121 | 1,192,320.9 | 3.0 | **** | 67.8 | 62.2 | 59.9 |
| FLE | 1 | 27.2 | 55.9 | **** | 121 | 4260.4 | 72.4 | **** | 121 | 217.2 | 3.7 | **** | 78.2 | 80.9 | 74.2 |
| FWI | 1 | 11.5 | 12.1 | *** | 121 | 8233.2 | 71.4 | **** | 121 | 344.9 | 3.0 | **** | 78.9 | 80.6 | 76.6 |
| FL/W | 1 | 1.8 | 126.9 | **** | 121 | 209.2 | 120.4 | **** | 121 | 4.4 | 2.5 | **** | 85.1 | 89.3 | 85.8 |
| NOL | 1 | 56.3 | 16.1 | **** | 121 | 30,730.5 | 72.5 | **** | 121 | 1009.8 | 2.4 | **** | 81.2 | 80.4 | 79.0 |
| PTK | 1 | 2.6 | 347.0 | **** | 121 | 26.7 | 29.1 | **** | 121 | 2.5 | 2.7 | **** | 55.8 | 70.5 | 58.8 |
| BRIX | 1 | 239.9 | 610.7 | **** | 121 | 798.1 | 16.8 | **** | 121 | 128.8 | 2.7 | **** | 44.5 | 55.5 | 42.3 |
| Group | Sample Size | Na | Ne | No. PA | He | uHe |
|---|---|---|---|---|---|---|
| ELR | 48 | 1.94 | 1.23 | 50 | 0.16 | 0.16 |
| SLR | 61 | 1.88 | 1.23 | 11 | 0.16 | 0.16 |
| CV | 11 | 1.92 | 1.44 | 1 | 0.28 | 0.29 |
| Overall | 120 | 2.00 | 1.25 | 0.18 | 0.18 |
| Comparison among Groups | ELR | SLR | CV |
|---|---|---|---|
| Genetic differentiation (FST) | |||
| ELR | *** | ** | |
| SLR | 0.04 | * | |
| CV | 0.09 | 0.08 | |
| Proportion of shared alleles | |||
| ELR | |||
| SLR | 0.93 | ||
| CV | 0.87 | 0.88 |
| Chromosome | Mean r2 | r2 Decay at 0.30 Mb | Mean rs2 | rs2 Decay at 0.22 Mb | Mean rv2 | rv2 Decay at 0.14 Mb | rvs2 | rvs2 Decay at 0.14 Mb |
|---|---|---|---|---|---|---|---|---|
| chr1 | 0.06 | 0.19 | 0.05 | 0.34 | 0.05 | 0.37 | 0.05 | 0.37 |
| chr2 | 0.14 | 0.63 | 0.11 | 0.91 | 0.11 | 1.02 | 0.11 | 0.94 |
| chr3 | 0.08 | 0.21 | 0.07 | 0.32 | 0.07 | 0.34 | 0.07 | 0.33 |
| chr4 | 0.13 | 0.70 | 0.11 | 1.13 | 0.11 | 0.80 | 0.11 | 0.81 |
| chr5 | 0.34 | 42.57 | 0.17 | 8.74 | 0.17 | 15.97 | 0.17 | 8.12 |
| chr6 | 0.09 | 0.35 | 0.09 | 0.69 | 0.09 | 0.83 | 0.09 | 0.84 |
| chr7 | 0.15 | 0.75 | 0.13 | 1.29 | 0.13 | 1.65 | 0.13 | 1.65 |
| chr8 | 0.06 | 0.12 | 0.05 | 0.21 | 0.05 | 0.27 | 0.05 | 0.27 |
| chr9 | 0.13 | 0.40 | 0.12 | 0.68 | 0.12 | 1.03 | 0.12 | 1.04 |
| chr10 | 0.08 | 0.09 | 0.07 | 0.17 | 0.07 | 0.25 | 0.07 | 0.25 |
| chr11 | 0.13 | 0.74 | 0.09 | 0.52 | 0.09 | 0.55 | 0.09 | 0.55 |
| chr12 | 0.24 | 2.11 | 0.23 | 3.63 | 0.23 | 1.36 | 0.23 | 1.37 |
| Mean | 0.15 | 0.63 | 0.11 | 0.81 | 0.07 | 0.80 | 0.07 | 0.80 |
| Method/Trait Type | CHR1 | CHR2 | CHR3 | CHR4 | CHR5 | CHR6 | CHR7 | CHR8 | CHR9 | CHR10 | CHR11 | CHR12 | TOTAL |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| FarmCPU | 30 | 30 | 36 | 26 | 22 | 17 | 8 | 17 | 9 | 19 | 44 | 7 | 265 |
| CLIMATE | 2 | 5 | 13 | 4 | 6 | 4 | 3 | 2 | 2 | 6 | 9 | 3 | 59 |
| FRUIT_QUALITY | 4 | 1 | 2 | 1 | 1 | 1 | 1 | 1 | 12 | ||||
| FRUIT_SHAPE_CP | 3 | 5 | 2 | 2 | 4 | 1 | 3 | 2 | 2 | 4 | 28 | ||
| FRUIT_SHAPE_DP | 6 | 6 | 8 | 13 | 6 | 2 | 1 | 9 | 3 | 2 | 15 | 3 | 74 |
| FRUIT_SIZE_CP | 2 | 3 | 4 | 1 | 2 | 1 | 2 | 3 | 18 | ||||
| FRUIT_SIZE_DP | 8 | 5 | 2 | 1 | 3 | 6 | 2 | 2 | 6 | 35 | |||
| GROWTH | 1 | 1 | 1 | 2 | 5 | ||||||||
| INFLORESCENCE | 2 | 1 | 1 | 1 | 1 | 2 | 3 | 1 | 12 | ||||
| LEAF TRAITS | 1 | 2 | 3 | 3 | 1 | 3 | 1 | 1 | 1 | 16 | |||
| PHENOLOGY | 1 | 2 | 1 | 1 | 1 | 6 | |||||||
| GAPIT.MLM | 5 | 4 | 3 | 6 | 5 | 2 | 6 | 8 | 1 | 40 | |||
| CLIMATE | 2 | 3 | 2 | 5 | 2 | 1 | 15 | ||||||
| FRUIT_QUALITY | 3 | 1 | 4 | ||||||||||
| FRUIT_SHAPE_CP | 3 | 1 | 1 | 4 | 9 | ||||||||
| FRUIT_SHAPE_DP | 1 | 3 | 1 | 3 | 8 | ||||||||
| FRUIT_SIZE_CP | 4 | 4 | |||||||||||
| QTCAT | 29 | 33 | 45 | 5 | 12 | 14 | 10 | 7 | 13 | 57 | 6 | 231 | |
| FRUIT_QUALITY | 1 | 1 | 1 | 2 | 1 | 6 | |||||||
| FRUIT_SHAPE_CP | 1 | 4 | 6 | 2 | 3 | 7 | 1 | 24 | |||||
| FRUIT_SHAPE_DP | 2 | 14 | 19 | 5 | 6 | 8 | 5 | 3 | 3 | 13 | 2 | 80 | |
| FRUIT_SIZE_CP | 7 | 2 | 6 | 1 | 3 | 4 | 3 | 26 | |||||
| FRUIT_SIZE_DP | 15 | 12 | 13 | 5 | 4 | 3 | 2 | 4 | 24 | 82 | |||
| INFLORESCENCE | 1 | 1 | 8 | 10 | |||||||||
| LEAF TRAITS | 1 | 1 | |||||||||||
| PHENOLOGY | 2 | 2 | |||||||||||
| TOTAL | 64 | 67 | 84 | 37 | 39 | 33 | 8 | 33 | 16 | 32 | 109 | 14 | 536 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rodriguez, M.; Scintu, A.; Posadinu, C.M.; Xu, Y.; Nguyen, C.V.; Sun, H.; Bitocchi, E.; Bellucci, E.; Papa, R.; Fei, Z.; et al. GWAS Based on RNA-Seq SNPs and High-Throughput Phenotyping Combined with Climatic Data Highlights the Reservoir of Valuable Genetic Diversity in Regional Tomato Landraces. Genes 2020, 11, 1387. https://doi.org/10.3390/genes11111387
Rodriguez M, Scintu A, Posadinu CM, Xu Y, Nguyen CV, Sun H, Bitocchi E, Bellucci E, Papa R, Fei Z, et al. GWAS Based on RNA-Seq SNPs and High-Throughput Phenotyping Combined with Climatic Data Highlights the Reservoir of Valuable Genetic Diversity in Regional Tomato Landraces. Genes. 2020; 11(11):1387. https://doi.org/10.3390/genes11111387
Chicago/Turabian StyleRodriguez, Monica, Alessandro Scintu, Chiara M. Posadinu, Yimin Xu, Cuong V. Nguyen, Honghe Sun, Elena Bitocchi, Elisa Bellucci, Roberto Papa, Zhangjun Fei, and et al. 2020. "GWAS Based on RNA-Seq SNPs and High-Throughput Phenotyping Combined with Climatic Data Highlights the Reservoir of Valuable Genetic Diversity in Regional Tomato Landraces" Genes 11, no. 11: 1387. https://doi.org/10.3390/genes11111387