1. Introduction
Advances in next-generation sequencing (NGS) technologies allow for efficient whole-mitochondrial-genome (mitogenome) sequence analysis of high-quality and degraded DNA samples [
1,
2,
3,
4]. NGS generates large amounts of data per sample and high read depth, which allows for increased sensitivity [
3,
5,
6]. The use of automated processing in NGS, especially for library preparation, reduces hands-on time and thus decreases the risk of human error (e.g., contamination and sample switches) [
7]. Additionally, the automated nature of NGS data analysis minimizes bias in data interpretation. Homopolymer stretches can be analyzed quantitatively to determine the major molecule within the sequence data, which is an improvement over the visual method applied to the capillary electrophoresis data produced from Sanger sequencing [
8,
9,
10]. The identification and removal of nuclear mitochondrial DNA segments (NUMTs) is also possible in NGS analysis using bioinformatic tools to investigate phasing of variants along parsed sequence reads and to perform multiple reference sequence alignment [
11,
12]. Automatic haplogrouping of complete mitogenome sequences is an additional quality control (QC) measure that can be used to flag unusual or phylogenetically implausible haplotypes arising from processing errors. Hence, NGS allows for more thorough QC of mitochondrial DNA (mtDNA) sequences than Sanger sequencing.
MtDNA data are numerous in the published literature, but sufficient quality-controlled mitogenome data for haplotype frequency estimations are lacking. The European DNA Profiling Group (EDNAP) mtDNA Population Database (EMPOP) is the preferred mtDNA haplotype database of the forensic community because it uses a robust QC pipeline to minimize the inclusion of error-ridden haplotypes [
13,
14]. At the time of writing, EMPOP contains only 4289 forensic-quality entire mitogenome profiles for haplotype frequency estimation. With the support of EMPOP and the Institut für Gerichtliche Medizin (GMI), the Armed Forces Medical Examiner System’s Armed Forces DNA Identification Laboratory (AFMES-AFDIL) was awarded a National Institute of Justice (NIJ) grant to augment the EMPOP database with mitogenome haplotypes from 4000 United States (U.S.) and 1000 global population samples. Many of the samples were previously sequenced in the control region (CR) with Sanger sequencing [
15,
16,
17,
18], allowing for comparison between the NGS and Sanger CR data and thus additional QC. In this report, the first completed datasets from the NIJ grant project are presented. As part of this effort, mitogenomes produced by the Applied Genetics group at the National Institute of Standards and Technology (NIST) were provided to the AFMES-AFDIL for analysis and inclusion in this study.
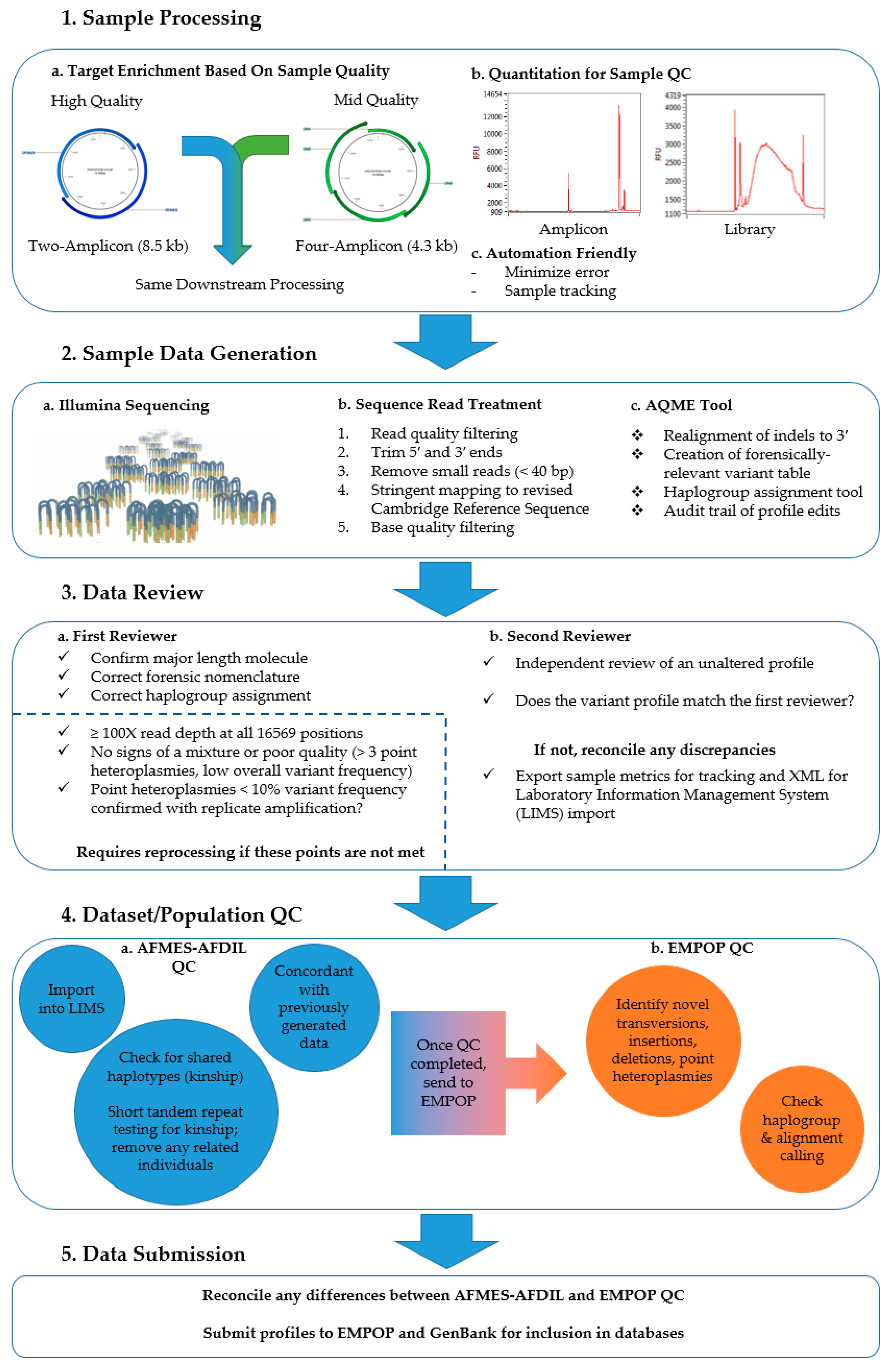
Over 1300 mtDNA profiles were generated using a thoroughly tested NGS processing method with automated data analysis and rigorous QC measures [
19,
20]. This method utilizes two-amplicon long-range PCR amplification, the KAPA HyperPlus Library Preparation Kit (Roche Sequencing, Waltham, MA, USA), and sequencing on the Illumina MiSeq (Illumina, San Diego, CA, USA). The two-amplicon approach minimizes the potential for co-amplification of nuclear mtDNA segments (NUMTs) that can complicate data analysis [
11,
21,
22]. The mitigation of NUMT interference with long-range enrichment allows for the use of a lower variant frequency threshold than short amplicon and whole-genome sequencing methods [
11,
23]. The library preparation method used in this study, KAPA HyperPlus, was shown to produce even coverage across the mitogenome to maximize sequencing efficiency [
19]. Furthermore, this library preparation method can accommodate a wide range of DNA input and does not require library amplification. KAPA HyperPlus therefore minimizes amplicon dilution prior to library preparation and eliminates the potential for PCR bias introduced during the library procedure [
19]. The Illumina MiSeq sequencing performed herein is advantageous due to its higher throughput and robust performance in homopolymer regions compared to pH-based sequencing [
9,
24]. Using an automated laboratory and analysis pipeline with a redundant data review, low-frequency variant confirmation through independent replication, and external EMPOP QC, ‘platinum-quality’ mitogenome haplotypes suitable for forensic use are provided.
4. Discussion
Forensic-quality mitogenomes were generated for 97% (1327/1365) of the samples attempted in this study. Despite slight differences in NGS processing (
Table 2), no loss of quality was observed in the data from any one method. Though the read count and read depth metrics differ between each data source, these metrics can be affected by a variety of factors such as the number of samples included in the MiSeq run, the read type (paired end v. single end), and the cluster density of the run. The major base frequency metrics, which are more indicative of sample data quality, were comparable between all three data sources, averaging > 99% when heteroplasmy was excluded. This indicates that minimal background noise (<1%) was observed in all datasets regardless of variations in sample type and processing. Thus, the two-amplicon long-range approach (supplemented with four-amplicon enrichment for lower quality samples), HyperPlus library preparation, and MiSeq sequencing, combined with the robust analysis pipeline, produced more than 1300 platinum-quality mitogenomes.
The four-amplicon enrichment approach was useful for processing samples that previously failed with the two-amplicon approach. However, NUMT interference was observed at 2–3% minor base frequency in the 4C amplicon of three samples. These NUMTs were below the 5% minor variant detection but emphasized the importance of data QC when analyzing at lower variant detection thresholds (<5%). It is also noteworthy that all three NUMTs were observed in the NIST samples extracted from whole blood, which has a higher likelihood for co-enrichment of NUMTs [
49]. However, no NUMTs were observed in data generated with the two-amplicon method, demonstrating the advantage of using the two-amplicon enrichment approach to generate reliable mitogenome data. The application of two-amplicon long-range enrichment allows primers to be specific for the mitogenome, opposed to small-amplicon approaches that may be homologous to regions of the nuclear genome due to primer design restrictions and mtDNA variation [
5,
11,
22,
50].
STR testing on samples with shared haplotypes was an important aspect of QC in this study, helping to identify duplicate or related samples for removal from the database. The inclusion of maternally related individuals in mtDNA databases can result in inaccurate estimation of haplotype frequencies [
37]. The use of both STR and SNP data allowed for the identification of two sets of more distant (second- and third-degree) relatives compared to traditional STR testing, which is typically only suitable for kinship analysis of first-degree relatives [
51,
52]. Since these more distant relatives were numbered sequentially in each case, it was presumed that the relatives were sampled simultaneously and that a genetic relationship was recognized between them. Other cases in which genetic relatives, recognized or not, were randomly sampled on separate occasions, would be better representative of the general population and might not be excluded from a mitogenome database. As new STR and SNP assays allow for more distant kinship testing [
53], it will be important to develop guidelines for the appropriate threshold for inclusion of relatives in population databases of lineage markers.
The automated nature of NGS data analysis combined with read count confirmation ensured that the major molecule was accurately assigned in homopolymer regions for all samples, in accordance with ISFG guidelines for population data [
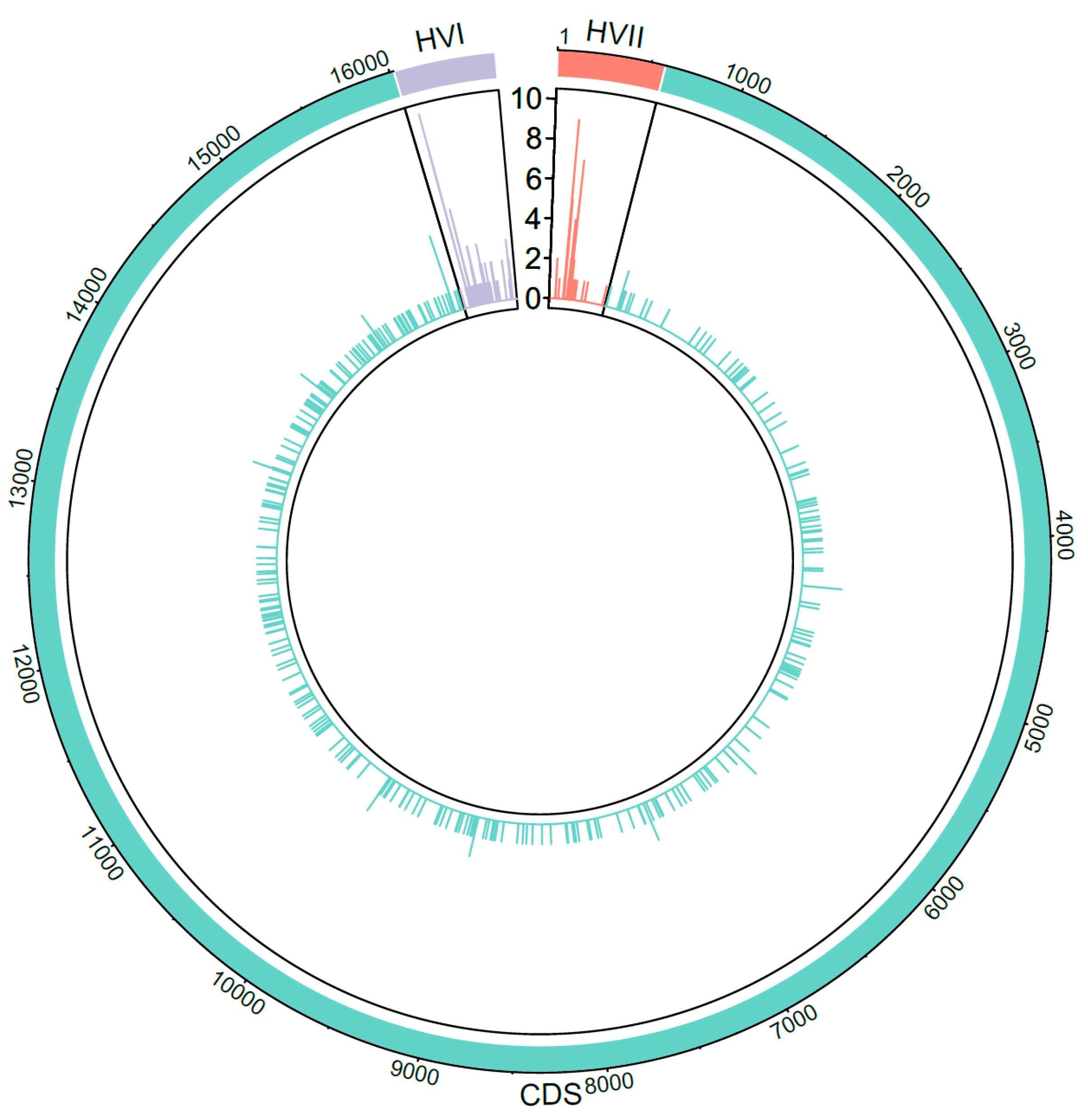
14]. The robust analysis workflow used in this study identified haplotypes with indels in common length heteroplasmy regions, which are recognized in EMPOP and can be disregarded in EMPOP haplotype queries. Indels were furthermore observed at eight additional CR and CDS sites that are not currently possible to ignore in EMPOP queries. However, these eight additional indels were observed at low frequency (<0.1% of haplotypes) with only one (nps 1878, 2141, 2417, 8289, 14,529, 15,545, 16,166, 16,296) or two (np 71) samples each. Because of their rarity, indels in these eight regions may not require special handling in database queries.
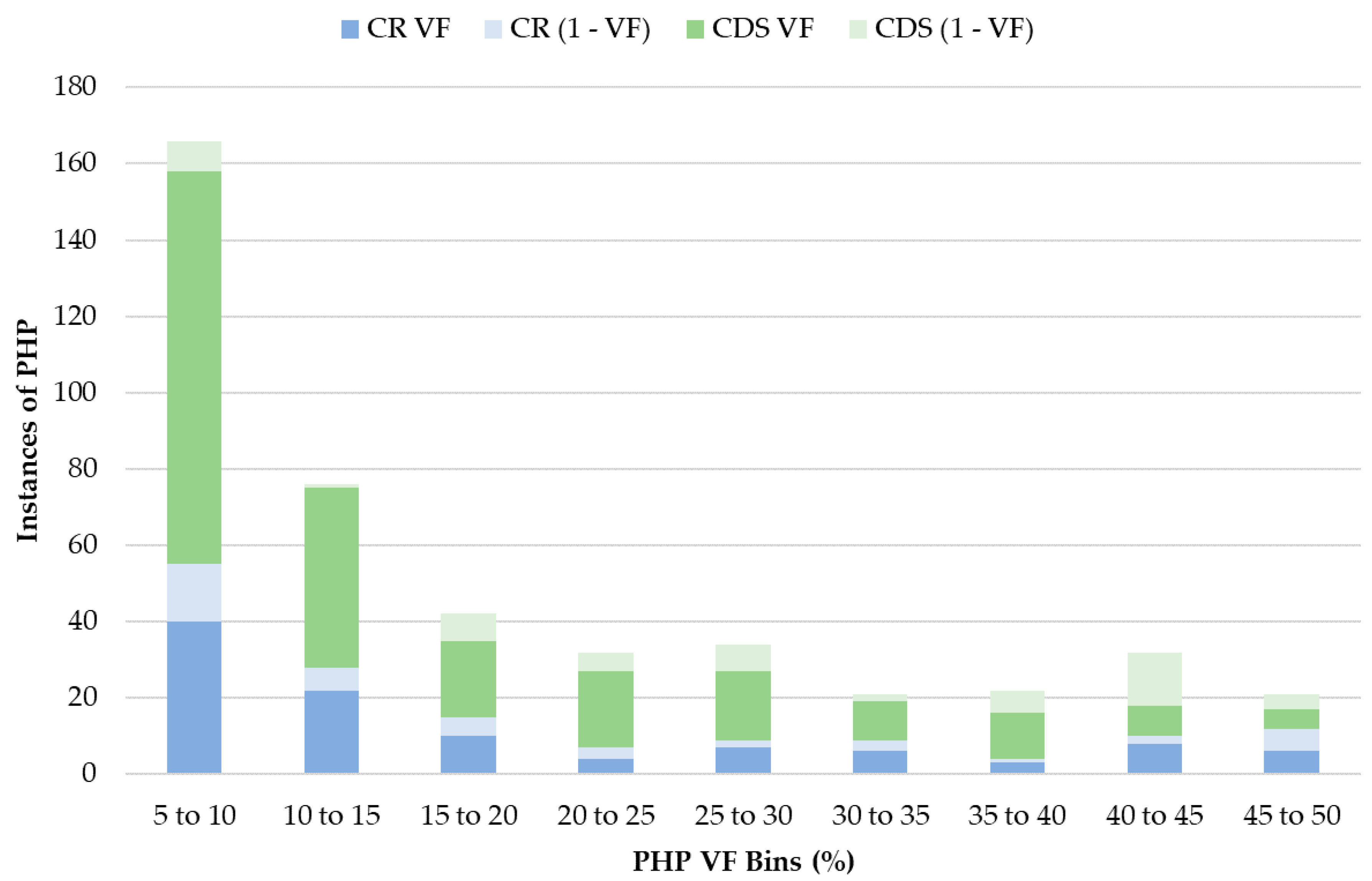
The authenticity of low-frequency PHPs presented in this study is ensured by the fact that every PHP with a minor base frequency < 10%, down to the minimum detection threshold of 5%, was confirmed in at least two separate amplification events. The importance of PHP confirmation for QC was emphasized by the fact that 22 PHPs could not be reproduced in the second amplification. Often in these cases, re-amplification would result in higher amplicon yields, but the PHP was not observed, indicating that the original observation was due to stochastic error. Inclusion of these spurious PHPs in the final datasets would over-represent mtDNA heteroplasmy rates and thus inflate haplotype diversity estimations [
54,
55,
56]. Of the reported PHPs, the majority (66.6%) were located in the CDS, consistent with the findings of [
57] in which 61.4% of observed PHPs were located in the CDS. In this study, PHPs were most frequently observed at CR positions 146, 152, 204, and 16,093, all of which are known PHP hotspots [
57,
58,
59,
60]. Unlike the findings in [
57,
58], not all CDS PHPs observed in this study were unique, with ten positions having more than one PHP occurrence. However, this difference is likely due to the larger sample size of the present study, which is one of the largest forensic mitogenome datasets published to date. Additionally, these previous studies utilized Sanger sequencing and thus were limited to a 10–20% threshold, the point at which when minor base signal could be confidently discerned from CE background noise. This is in contrast to the 5% heteroplasmy detection threshold used in this study, which can reveal lower-level variants than Sanger sequencing. Finally, the majority (95.1%) of the reported PHPs were transition-type, consistent with findings in [
57], in which 96% of PHPs were transitions.
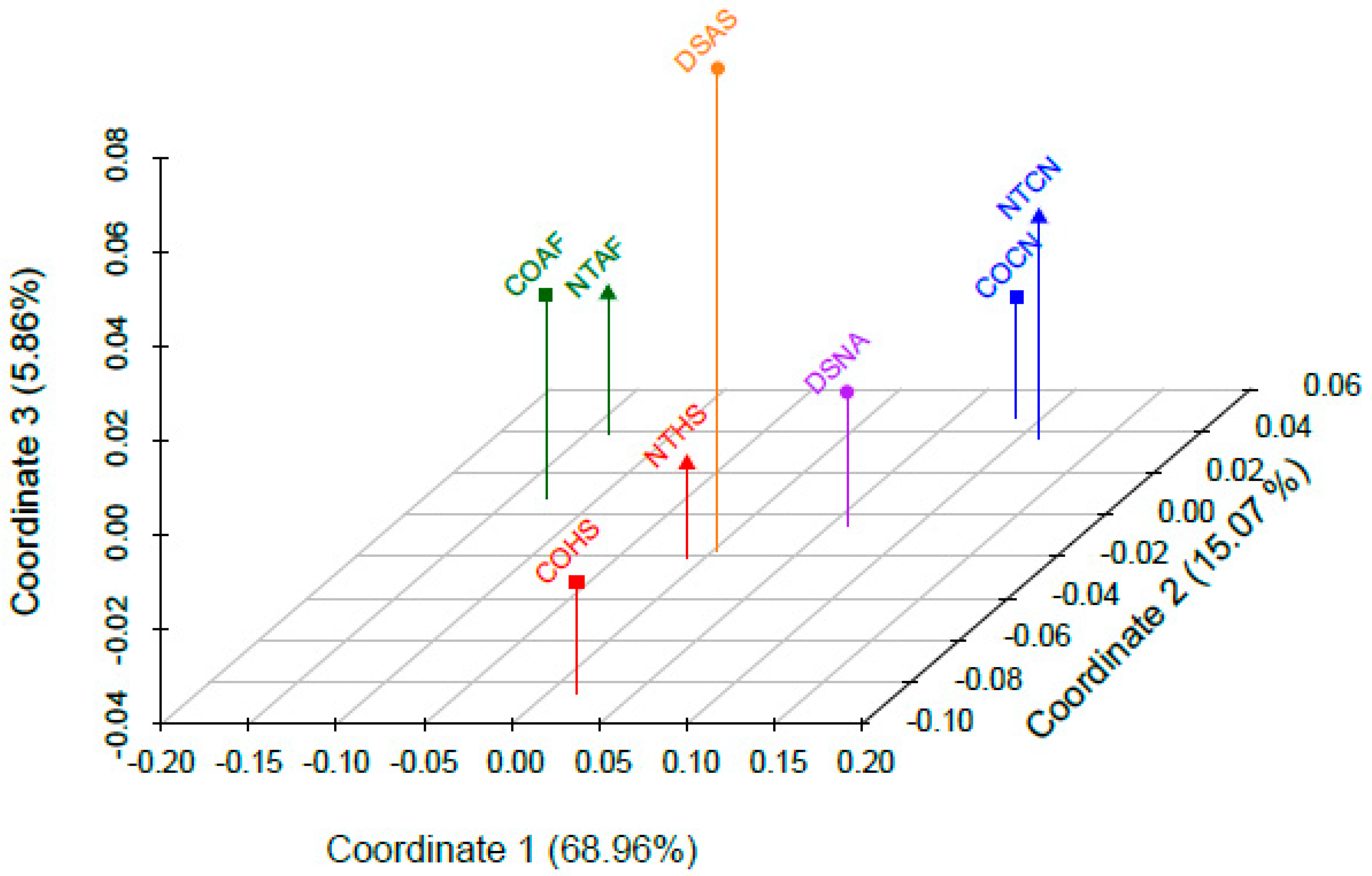
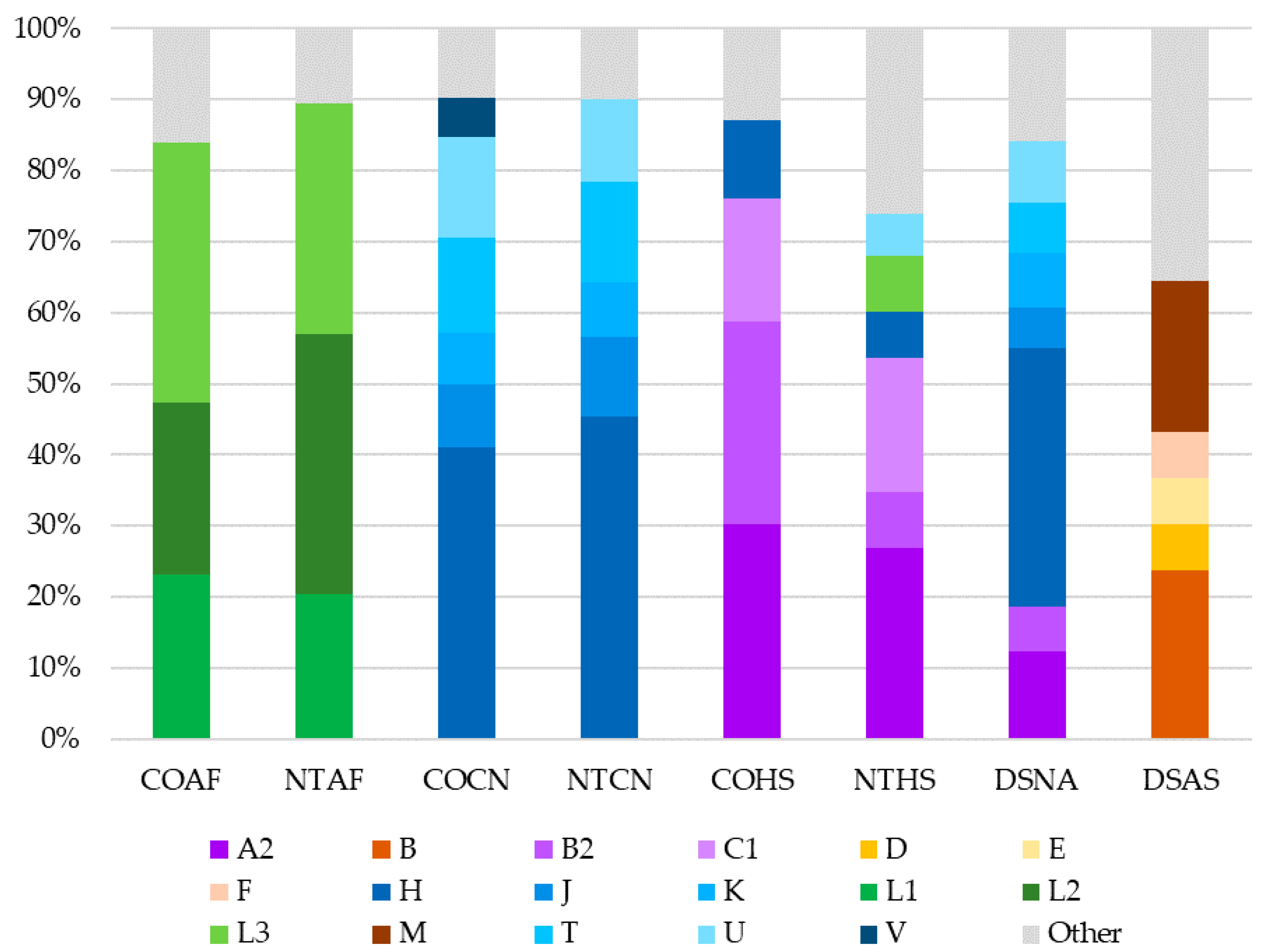
When pairwise Fst values were used to estimate genetic distance between the eight datasets, it was found that all comparisons except for those between the two African American (COAF and NTAF) and two Caucasian (COCN and NTCN) datasets were statistically significantly different from one another. Based on these findings, it would be reasonable for a laboratory to combine the African American and Caucasian datasets to increase sample size when conducting haplotype match statistics. Conversely, the difference between the two Hispanic (COHS and NTHS) datasets was noteworthy. Though the largest proportions of haplogroups for both the COHS and NTHS Hispanic datasets were of Native American ancestry (80.7% and 56.5%, respectively), the NTHS had a much larger proportion of European and African lineages (42% total) compared to the COHS dataset (18.3% total). The difference in haplogroup distribution between these two Hispanic datasets is likely due to the differing geographic origin of each sample set, as the NTHS samples were collected in the northeast and southern United States whereas the COHS samples were mostly from Colorado. Based on the 2010 U.S. Census, Hispanics from the western United States (including Colorado) are predominantly from Mexico and Central America, whereas a large proportion of Hispanics in the northeast and Florida originate from the Caribbean [
61]. As a result, the differing population structure of the Central American and Caribbean parental populations likely contributed to the observed differences between the two Hispanic datasets. In particular, the greater proportion of African ancestry in Caribbean populations [
62] may explain the prevalence of African lineages in the NTHS dataset [
25]. The geographic sampling differences between the Hispanic datasets is furthermore evident in their haplogroup B2 proportions. Haplogroup B2 is a Native American lineage found in relatively high frequencies in the southwestern U.S. [
63,
64,
65]. Therefore, the proportion of B2 haplotypes in the COHS dataset, which includes individuals primarily from Colorado (a southwestern state), was greater than that of the NTHS dataset originating from eastern and southern states. These analyses highlight the distinctiveness of the two geographically disparate Hispanic mitogenome datasets included in this study.
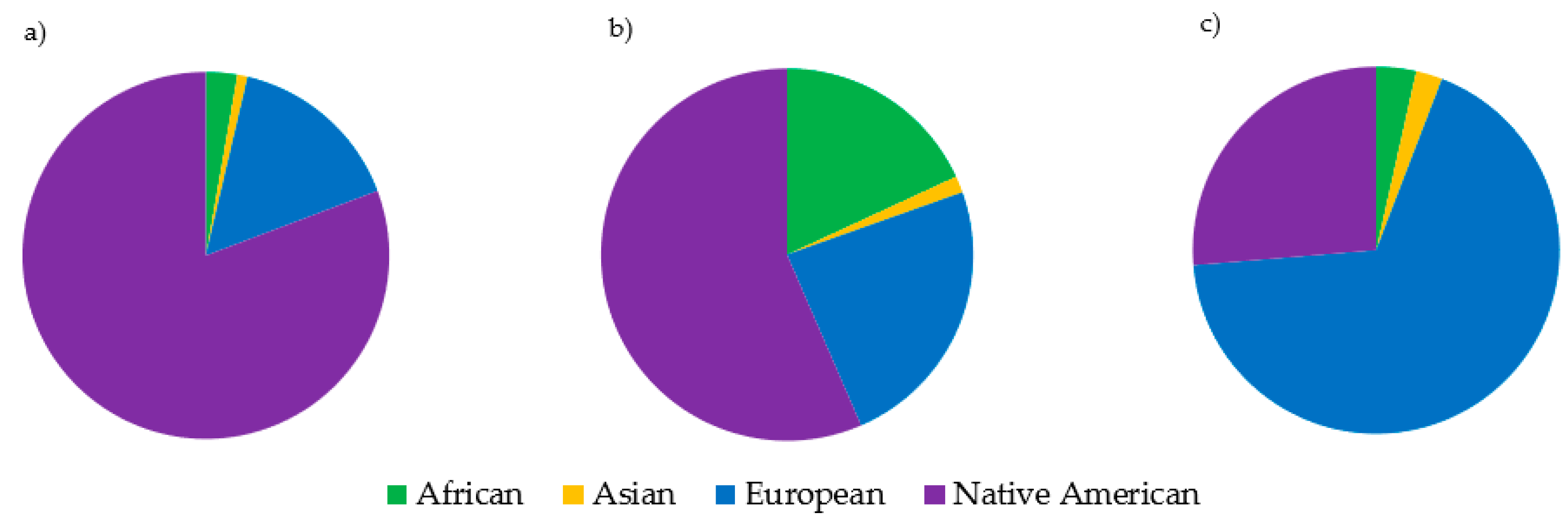
It was not unexpected that the Asian (DSAS) and Native American (DSNA) datasets were different from the other datasets (and each other), given that there was only one dataset per metapopulation. The mtDNA haplogroup distribution observed in the Native American (DSNA) dataset was interesting, with 67% of haplotypes belonging to European haplogroups. This finding explains why the Native American dataset plotted between the Caucasian and Hispanic datasets, considering the high proportion of Native American haplotypes in the Hispanic datasets. The finding that the Native American dataset contained a lower proportion of Native American haplotypes than the Hispanic datasets underscores the complexity behind self-identification as it relates to DNA and genetic ancestry. This demonstrates that mitochondrial ancestry may be inconsistent with expectations based on the self-reported metapopulation, especially for admixed and indigenous groups.

 ,
, 






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}