Metabolic Specialization and Codon Preference of Lignocellulolytic Genes in the White Rot Basidiomycete Ceriporiopsis subvermispora



Abstract
:1. Introduction
2. Materials and Methods
2.1. Sequences
2.2. Microarray Data
2.3. Determination of the Gene Copy Number Coding for tRNAs
2.4. Determination of Codons and Codon Pair Frequencies
2.5. Determination of Bias in Codon-Pair Usage
2.6. Calculation of Relative Synonymous Codon Usage (RSCU) and Codon Adaptation Index (CAI)
2.7. Determination of Adaptation to the tRNA Pool
2.8. Determination of AAtAI
2.9. Determination of effective Number of Codons (Nc)
2.10. Phylogenetic Analysis
2.11. Graphs and Statistical Methods
3. Results
3.1. C. subvermispora tRNAs
3.2. Phylogenetic Analysis of C. subvermispora tRNA Genes
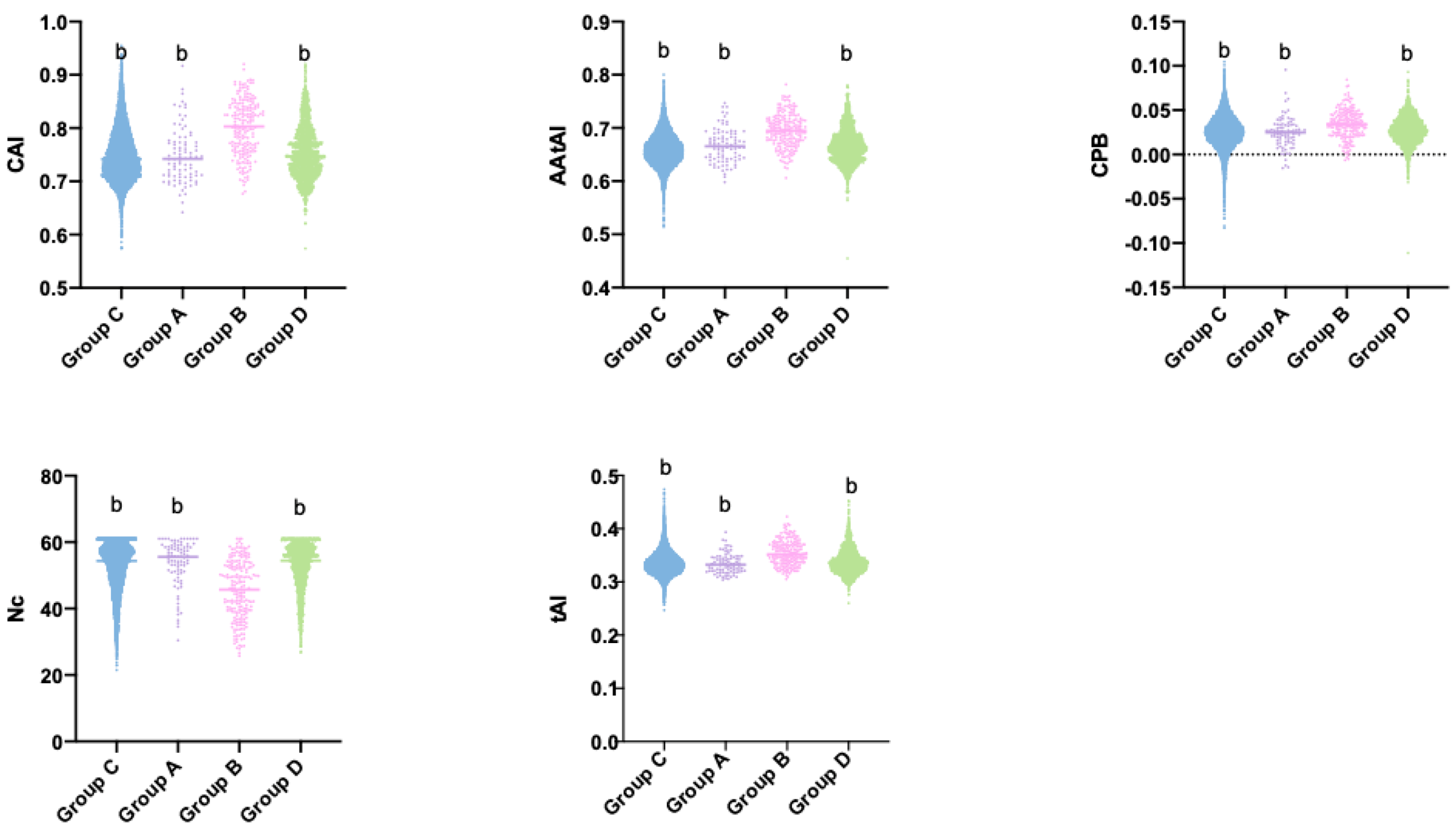
3.3. tRNA Abundance and Codon Usage in C. subvermispora
3.4. Relationship between Gene Expression Level, Codon Bias, and Translational Efficiency in C. subvermispora
3.5. Transcriptional Response to Growth on Ball-Milled Aspen (BMA), Codon Bias, and Translational Efficiency
3.6. Translational Efficiency and Codon Bias in Lignocellulolytic Genes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Martínez, A.T.; Speranza, M.; Ruiz-Dueñas, F.J.; Ferreira, P.; Camarero, S.; Guillén, F.; Martínez, M.J.; Gutiérrez, A.; del Río, J.C. Biodegradation of lignocellulosics: Microbial, chemical, and enzymatic aspects of the fungal attack of lignin. Int. Microbiol. 2005, 8, 195–204. [Google Scholar] [PubMed]
- Kersten, P.; Cullen, D. Extracellular oxidative systems of the lignin-degrading Basidiomycete Phanerochaete chrysosporium. Fungal Genet. Biol. 2007, 44, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Wan, C.; Li, Y. Fungal pretreatment of lignocellulosic biomass. Biotechnol. Adv. 2012, 30, 1447–1457. [Google Scholar] [CrossRef]
- Baldrian, P.; Valásková, V. Degradation of cellulose by basidiomycetous fungi. FEMS Microbiol. Rev. 2008, 32, 501–521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rüttimann-Johnson, C.; Salas, L.; Vicuña, R.; Kirk, T.K. Extracellular Enzyme Production and Synthetic Lignin Mineralization by Ceriporiopsis subvermispora. Appl. Environ. Microbiol. 1993, 59, 1792–1797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lobos, S.; Larraín, J.; Salas, L.; Cullen, D.; Vicuña, R. Isoenzymes of manganese-dependent peroxidase and laccase produced by the lignin-degrading basidiomycete Ceriporiopsis subvermispora. Microbiology 1994, 140, 2691–2698. [Google Scholar] [CrossRef] [Green Version]
- Enoki, M.; Watanabe, T.; Nakagame, S.; Koller, K.; Messner, K.; Honda, Y.; Kuwahara, M. Extracellular lipid peroxidation of selective white-rot fungus, Ceriporiopsis subvermispora. FEMS Microbiol. Lett. 1999, 180, 205–211. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Fueyo, E.; Ruiz-Dueñas, F.J.; Ferreira, P.; Floudas, D.; Hibbett, D.S.; Canessa, P.; Larrondo, L.F.; James, T.Y.; Seelenfreund, D.; Lobos, S.; et al. Comparative genomics of Ceriporiopsis subvermispora and Phanerochaete chrysosporium provide insight into selective ligninolysis. Proc. Natl. Acad. Sci. USA 2012, 109, 5458–5463. [Google Scholar] [CrossRef] [Green Version]
- Fernández-Fueyo, E.; Ruiz-Dueñas, F.J.; Miki, Y.; Martínez, M.J.; Hammel, K.E.; Martínez, A.T. Lignin-degrading peroxidases from genome of selective ligninolytic fungus Ceriporiopsis subvermispora. J. Biol. Chem. 2012, 287, 16903–16916. [Google Scholar] [CrossRef] [Green Version]
- Salas, C.; Lobos, S.; Larraín, J.; Salas, L.; Cullen, D.; Vicuña, R. Properties of laccase isoenzymes produced by the basidiomycete Ceriporiopsis subvermispora. Biotechnol. Appl. Biochem. 1995, 21, 323–333. [Google Scholar]
- Tello, M.; Corsini, G.; Larrondo, L.F.; Salas, L.; Lobos, S.; Vicuña, R. Characterization of three new manganese peroxidase genes from the ligninolytic basidiomycete Ceriporiopsis subvermispora. Biochim. Biophys. Acta 2000, 1490, 137–144. [Google Scholar] [CrossRef]
- Tello, M.; Seelenfreund, D.; Lobos, S.; Gaskell, J.; Cullen, D.; Vicuña, R. Isolation and characterization of homokaryotic strains from the ligninolytic basidiomycete Ceriporiopsis subvermispora. FEMS Microbiol. Lett. 2001, 199, 91–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manubens, A.; Canessa, P.; Folch, C.; Avila, M.; Salas, L.; Vicuña, R. Manganese affects the production of laccase in the basidiomycete Ceriporiopsis subvermispora. FEMS Microbiol. Lett. 2007, 275, 139–145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gutiérrez, M.; Rojas, L.A.; Mancilla-Villalobos, R.; Seelenfreund, D.; Vicuña, R.; Lobos, S. Analysis of manganese-regulated gene expression in the ligninolytic basidiomycete Ceriporiopsis subvermispora. Curr. Genet. 2008, 54, 163–173. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, J.M.; Canessa, P.; Mancilla, R.A.; Polanco, R.; Santibáñez, P.A.; Vicuña, R. Expression of genes encoding laccase and manganese-dependent peroxidase in the fungus Ceriporiopsis subvermispora is mediated by an ACE1-like copper-fist transcription factor. Fungal Genet. Biol. 2009, 46, 104–111. [Google Scholar] [CrossRef] [PubMed]
- Mancilla, R.A.; Canessa, P.; Manubens, A.; Vicuña, R. Effect of manganese on the secretion of manganese-peroxidase by the basidiomycete Ceriporiopsis subvermispora. Fungal Genet. Biol. 2010, 47, 656–661. [Google Scholar] [CrossRef] [PubMed]
- Hori, C.; Gaskell, J.; Igarashi, K.; Kersten, P.; Mozuch, M.; Samejima, M.; Cullen, D. Temporal alterations in the secretome of the selective ligninolytic fungus Ceriporiopsis subvermispora during growth on aspen wood reveal this organism’s strategy for degrading lignocellulose. Appl. Environ. Microbiol. 2014, 80, 2062–2070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manubens, A.; Avila, M.; Canessa, P.; Vicuña, R. Differential regulation of genes encoding manganese peroxidase (MnP) in the basidiomycete Ceriporiopsis subvermispora. Curr. Genet. 2003, 43, 433–438. [Google Scholar] [CrossRef]
- Sharp, P.M.; Stenico, M.; Peden, J.F.; Lloyd, A.T. Codon usage: Mutational bias, translational selection, or both? Biochem. Soc. Trans. 1993, 21, 835–841. [Google Scholar] [CrossRef] [Green Version]
- Supek, F.; Skunca, N.; Repar, J.; Vlahovicek, K.; Smuc, T. Translational selection is ubiquitous in prokaryotes. PLoS Genet. 2010, 6, e1001004. [Google Scholar] [CrossRef] [Green Version]
- Tuller, T.; Waldman, Y.Y.; Kupiec, M.; Ruppin, E. Translation efficiency is determined by both codon bias and folding energy. Proc. Natl. Acad. Sci. USA 2010, 107, 3645–3650. [Google Scholar] [CrossRef] [Green Version]
- Sharp, P.M.; Li, W.H. The codon Adaptation Index--a measure of directional synonymous codon usage bias, and its potential applications. Nucleic Acids Res. 1987, 15, 1281–1295. [Google Scholar] [CrossRef] [Green Version]
- Bulmer, M. Coevolution of codon usage and transfer RNA abundance. Nature 1987, 325, 728–730. [Google Scholar] [CrossRef] [PubMed]
- dos Reis, M.; Savva, R.; Wernisch, L. Solving the riddle of codon usage preferences: A test for translational selection. Nucleic Acids Res. 2004, 32, 5036–5044. [Google Scholar] [CrossRef] [Green Version]
- Lowe, T.M.; Eddy, S.R. tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997, 25, 955–964. [Google Scholar] [CrossRef] [PubMed]
- Carver, T.; Bleasby, A. The design of Jemboss: A graphical user interface to EMBOSS. Bioinformatics 2003, 19, 1837–1843. [Google Scholar] [CrossRef]
- Tello, M.; Saavedra, J.M.; Spencer, E. Analysis of the use of codon pairs in the HE gene of the ISA virus shows a correlation between bias in HPR codon-pair use and mortality rates caused by the virus. Virol. J. 2013, 10, 180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coleman, J.R.; Papamichail, D.; Skiena, S.; Futcher, B.; Wimmer, E.; Mueller, S. Virus attenuation by genome-scale changes in codon pair bias. Science 2008, 320, 1784–1787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mueller, S.; Papamichail, D.; Coleman, J.R.; Skiena, S.; Wimmer, E. Reduction of the rate of poliovirus protein synthesis through large-scale codon deoptimization causes attenuation of viral virulence by lowering specific infectivity. J. Virol. 2006, 80, 9687–9696. [Google Scholar] [CrossRef] [Green Version]
- Rice, P.; Longden, I.; Bleasby, A. EMBOSS: The European Molecular Biology Open Software Suite. Trends Genet. 2000, 16, 276–277. [Google Scholar] [CrossRef]
- Tuller, T.; Carmi, A.; Vestsigian, K.; Navon, S.; Dorfan, Y.; Zaborske, J.; Pan, T.; Dahan, O.; Furman, I.; Pilpel, Y. An evolutionarily conserved mechanism for controlling the efficiency of protein translation. Cell 2010, 141, 344–354. [Google Scholar] [CrossRef] [Green Version]
- Thompson, J.D.; Higgins, D.G.; Gibson, T.J. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994, 22, 4673–4680. [Google Scholar] [CrossRef] [Green Version]
- Saitou, N.; Nei, M. The neighbor-joining method: A new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 1987, 4, 406–425. [Google Scholar] [PubMed]
- Dopazo, J. Estimating errors and confidence intervals for branch lengths in phylogenetic trees by a bootstrap approach. J. Mol. Evol. 1994, 38, 300–304. [Google Scholar] [CrossRef]
- Tamura, K.; Nei, M.; Kumar, S. Prospects for inferring very large phylogenies by using the neighbor-joining method. Proc. Natl. Acad. Sci. USA 2004, 101, 11030–11035. [Google Scholar] [CrossRef] [Green Version]
- Tamura, K.; Peterson, D.; Peterson, N.; Stecher, G.; Nei, M.; Kumar, S. MEGA5: Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 2011, 28, 2731–2739. [Google Scholar] [CrossRef] [Green Version]
- Fingerman, I.; Nagaraj, V.; Norris, D.; Vershon, A.K. Sfp1 plays a key role in yeast ribosome biogenesis. Eukaryot. Cell 2003, 2, 1061–1068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marion, R.M.; Regev, A.; Segal, E.; Barash, Y.; Koller, D.; Friedman, N.; O’Shea, E.K. Sfp1 is a stress- and nutrient-sensitive regulator of ribosomal protein gene expression. Proc. Natl. Acad. Sci. USA 2004, 101, 14315–14322. [Google Scholar] [CrossRef] [Green Version]
- Tello, M.; Vergara, F.; Spencer, E. Genomic adaptation of the ISA virus to Salmo salar codon usage. Virol. J. 2013, 10, 223. [Google Scholar] [CrossRef] [Green Version]
- Zhao, V.; Jacobs, W.M.; Shakhnovich, E.I. Effect of protein structure on evolution of cotranslational folding. Biophys. J. 2020. [Google Scholar] [CrossRef] [PubMed]
- Bitran, A.; Jacobs, W.M.; Zhai, X.; Shakhnovich, E. Cotranslational folding allows misfolding-prone proteins to circumvent deep kinetic traps. Proc. Natl. Acad. Sci. USA 2020, 117, 1485–1495. [Google Scholar] [CrossRef]
- Aragonès, L.; Guix, S.; Ribes, E.; Bosch, A.; Pintó, R.M. Fine-tuning translation kinetics selection as the driving force of codon usage bias in the hepatitis A virus capsid. PLoS Pathog. 2010, 6, e1000797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plotkin, J.B.; Kudla, G. Synonymous but not the same: The causes and consequences of codon bias. Nat. Rev. Genet. 2011, 12, 32–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharp, P.M.; Tuohy, T.M.; Mosurski, K.R. Codon usage in yeast: Cluster analysis clearly differentiates highly and lowly expressed genes. Nucleic Acids Res. 1986, 14, 5125–5143. [Google Scholar] [CrossRef] [PubMed]
- Sharp, P.M.; Cowe, E.; Higgins, D.G.; Shields, D.C.; Wolfe, K.H.; Wright, F. Codon usage patterns in Escherichia coli, Bacillus subtilis, Saccharomyces cerevisiae, Schizosaccharomyces pombe, Drosophila melanogaster and Homo sapiens; a review of the considerable within-species diversity. Nucleic Acids Res. 1988, 16, 8207–8211. [Google Scholar] [CrossRef] [Green Version]
- Bahir, I.; Fromer, M.; Prat, Y.; Linial, M. Viral adaptation to host: A proteome-based analysis of codon usage and amino acid preferences. Mol. Syst. Biol. 2009, 5, 311. [Google Scholar] [CrossRef] [PubMed]
- Angov, E. Codon usage: Nature’s roadmap to expression and folding of proteins. Biotechnol. J. 2011, 6, 650–659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mueller, S.; Coleman, J.R.; Papamichail, D.; Ward, C.B.; Nimnual, A.; Futcher, B.; Skiena, S.; Wimmer, E. Live attenuated influenza virus vaccines by computer-aided rational design. Nat. Biotechnol. 2010, 28, 723–726. [Google Scholar] [CrossRef] [Green Version]
- Zhou, M.; Guo, J.; Cha, J.; Chae, M.; Chen, S.; Barral, J.M.; Sachs, M.S.; Liu, Y. Non-optimal codon usage affects expression, structure and function of clock protein FRQ. Nature 2013, 495, 111–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Novoa, E.M.; Pavon-Eternod, M.; Pan, T.; de Pouplana, L.R. A role for tRNA modifications in genome structure and codon usage. Cell 2012, 149, 202–213. [Google Scholar] [CrossRef] [Green Version]
- Wenke, T.; Döbel, T.; Sörensen, T.R.; Junghans, H.; Weisshaar, B.; Schmidt, T. Targeted identification of short interspersed nuclear element families shows their widespread existence and extreme heterogeneity in plant genomes. Plant Cell 2011, 23, 3117–3128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Enav, H.; Béjà, O.; Mandel-Gutfreund, Y. Cyanophage tRNAs may have a role in cross-infectivity of oceanic Prochlorococcus and Synechococcus hosts. ISME J. 2012, 6, 619–628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Weringh, A.; Ragonnet-Cronin, M.; Pranckeviciene, E.; Pavon-Eternod, M.; Kleiman, L.; Xia, X. HIV-1 modulates the tRNA pool to improve translation efficiency. Mol. Biol. Evol. 2011, 28, 1827–1834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deswal, D.; Khasa, Y.P.; Kuhad, R.C. Optimization of cellulase production by a brown rot fungus Fomitopsis sp. RCK2010 under solid state fermentation. Bioresour. Technol. 2011, 102, 6065–6072. [Google Scholar] [CrossRef] [PubMed]
- Levin, L.; Melignani, E.; Ramos, A.M. Effect of nitrogen sources and vitamins on ligninolytic enzyme production by some white-rot fungi. Dye decolorization by selected culture filtrates. Bioresour. Technol. 2010, 101, 4554–4563. [Google Scholar] [CrossRef]
- Heinemann, I.U.; Jahn, M.; Jahn, D. The biochemistry of heme biosynthesis. Arch. Biochem. Biophys. 2008, 474, 238–251. [Google Scholar] [CrossRef]
- Ritch, T.G.; Gold, M.H. Characterization of a highly expressed lignin peroxidase-encoding gene from the basidiomycete Phanerochaete chrysosporium. Gene 1992, 118, 73–80. [Google Scholar] [CrossRef]
- Floudas, D.; Binder, M.; Riley, R.; Barry, K.; Blanchette, R.A.; Henrissat, B.; Martínez, A.T.; Otillar, R.; Spatafora, J.W.; Yadav, J.S.; et al. The Paleozoic origin of enzymatic lignin decomposition reconstructed from 31 fungal genomes. Science 2012, 336, 1715–1719. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| tRNA Type | Number of tRNAs | Anticodon and Frequency * | Scaffolds | Number of tRNAs with Introns |
|---|---|---|---|---|
| Ala | 14 | AGC7, CGC5, TGC2 | 3, 5, 7, 12, 14, 18, 19 | 13 |
| Arg | 16 | ACG8, CCG2, CCT2, TCG3, TCT1 | 2, 3, 5, 9, 14, 19, 31, 39 | 15 |
| Asn | 5 | GTT5 | 4, 5, 7, 13, 14 | 5 |
| Asp | 10 | GTC10 | 1, 6, 8, 12, 16, 19, 28, 41 | 7 |
| Cys | 3 | GCA3 | 10, 13, 20 | 1 |
| Gln | 8 | CTG6, TTG2 | 1, 2, 6, 17, 18, 28 | 8 |
| Glu | 15 | CTC12, TTC3 | 3, 8, 9, 19, 28 | 5 |
| Gly | 17 | CCC2, GCC12, TCC3 | 1, 2, 5, 7, 9, 10, 13, 20 | 1 |
| His | 5 | GTG5 | 2, 6, 7, 12 | 5 |
| Ile | 9 | AAT8, TAT1 | 4, 5, 21, 33 | 9 |
| Leu | 15 | AAG7, CAA2, CAG4, TAA1, TAG1 | 1, 2, 4, 5, 6, 7, 9, 13, 17, 25, 27, 30 | 14 |
| Lys | 11 | CTT10, TTT1 | 1, 3 | 11 |
| Met | 8 | CAT8 | 1, 2, 3, 9, 10, 11, 12, 23 | 7 |
| Phe | 5 | GAA5 | 1, 5, 9, 10 | 4 |
| Pro | 10 | AGG5, CGG4, TGG1 | 4, 6, 7, 14, 18, 22, 35 | 10 |
| Ser | 13 | ACT1, AGA4, CGA3, GCT3, TGA2 | 1, 3, 4, 5, 6, 9, 15, 18 | 8 |
| Thr | 9 | AGT6, CGT2, TGT1 | 1, 3, 4, 8, 9, 10, 22 | 6 |
| Trp | 3 | CCA3 | 19, 20, 30 | 3 |
| Tyr | 4 | GTA4 | 2, 3, 8, 30 | 4 |
| Val | 11 | TAC1, CAC2, AAC6, CAC1, AAC1 | 5, 6, 12, 14, 15, 28, 32 | 2 |
| BMA (ρ) | Glu (ρ) | BMA/Glu (ρ) | BMA/Glu (ρ) a | BMA/Glu (ρ) b | BMA/Glu (ρ) c | |
|---|---|---|---|---|---|---|
| CAI | −4.95 × 10−3 | −8.04 × 10−2 *** | 3.39 × 10−1 *** | 4.20 × 10−1 ** | 4.66 × 10−1 *** | 3.09 × 10−1 *** |
| CPB | 6.09 × 10−2 *** | 6.59 × 10−3 *** | 2.61 × 10−1 *** | NA | 3.26 × 10−1 *** | 2.47 × 10−1 *** |
| Nc | 1.81 × 10−2 * | 8.17 × 10−2 *** | −2.63 × 10−1 *** | −4.51 × 10−1 *** | −4.42 × 10−1 *** | −2.21 × 10−1 *** |
| tAI | 3.48 × 10−2 *** | −3.52 × 10−2 *** | 3.17 × 10−1 *** | 2.84 × 10−1 * | 4.36 × 10−1 *** | 2.82 × 10−1 *** |
| AAtAI | 6.54 × 10−2 *** | 8.34 × 10−4 | 2.67 × 10−1 *** | 4.75 × 10−1 *** | 4.17 × 10−1 *** | 2.27 × 10−1 *** |
| Ligno-Cellulolytic Function | Transcript ID | CAI | AAtAI | tAI | CPB | Nc | Z-CAI | Z-AAtAI | Z-CPB | Z-tAI | Putative Function | Microarray Signal (log2) | |||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Glucose | BMA | BMA/Glu | p-Value | ||||||||||||
| Fungal lignin peroxidase | 49863 | 0.814 | 0.720 | 0.371 | 0.028 | 40.95 | 1.236 | 1.848 | 0.136 | 1.513 | Peroxidase, MnP | 9.58 | 11.15 | 2.970 | 0.08320 |
| 50297 | 0.822 | 0.735 | 0.384 | 0.055 | 41.46 | 1.394 | 2.321 | 1.565 | 2.111 | Peroxidase, MnP | 13.13 | 11.88 | 0.421 | 0.19000 | |
| 50686 | 0.823 | 0.728 | 0.379 | 0.051 | 41.50 | 1.414 | 2.100 | 1.387 | 1.901 | Peroxidase, MnP | 9.35 | 9.42 | 1.048 | 0.45600 | |
| 106380 | 0.815 | 0.716 | 0.371 | 0.039 | 38.67 | 1.255 | 1.722 | 0.705 | 1.536 | Peroxidase, MnP | 8.93 | 8.93 | 1.000 | 0.99900 | |
| 111364 | 0.770 | 0.689 | 0.357 | 0.015 | 50.57 | 0.364 | 0.872 | −0.611 | 0.929 | Peroxidase, VP-like | 9.18 | 9.11 | 0.957 | 0.52800 | |
| 117521 | 0.819 | 0.722 | 0.367 | 0.042 | 40.88 | 1.335 | 1.911 | 0.876 | 1.340 | Peroxidase, MnP | 9.57 | 9.49 | 0.945 | 0.30600 | |
| 124144 | 0.696 | 0.653 | 0.321 | 0.031 | 61.00 | −1.101 | −0.261 | 0.284 | −0.660 | Peroxidase, generic | 11.30 | 11.21 | 0.942 | 0.29300 | |
| 126018 | 0.828 | 0.721 | 0.366 | 0.030 | 40.55 | 1.513 | 1.880 | 0.230 | 1.314 | Peroxidase, MnP | 9.69 | 9.50 | 0.874 | 0.10000 | |
| 126058 | 0.788 | 0.673 | 0.344 | 0.026 | 44.68 | 0.721 | 0.368 | −0.015 | 0.340 | Peroxidase, MnP | 9.94 | 9.44 | 0.707 | 0.00595 | |
| 128590 | 0.824 | 0.724 | 0.367 | 0.052 | 41.12 | 1.434 | 1.974 | 1.409 | 1.352 | Peroxidase, MnP | 9.78 | 9.52 | 0.837 | 0.01960 | |
| 129418 | 0.767 | 0.709 | 0.373 | 0.044 | 49.06 | 0.305 | 1.502 | 1.011 | 1.619 | Peroxidase, MnP | 10.22 | 12.92 | 6.508 | 0.00895 | |
| 130659 | 0.781 | 0.721 | 0.368 | 0.022 | 50.53 | 0.582 | 1.880 | −0.220 | 1.396 | Peroxidase, LiP-like | 10.88 | 10.30 | 0.673 | 0.03900 | |
| 136058 | 0.770 | 0.688 | 0.352 | 0.029 | 50.91 | 0.364 | 0.841 | 0.167 | 0.683 | Peroxidase, MnP | 10.16 | 10.76 | 1.519 | 0.16700 | |
| 151947 | 0.831 | 0.729 | 0.373 | 0.052 | 40.42 | 1.572 | 2.132 | 1.410 | 1.600 | Peroxidase, MnP | 9.00 | 8.93 | 0.955 | 0.24200 | |
| 155372 | 0.797 | 0.711 | 0.369 | 0.040 | 42.88 | 0.899 | 1.565 | 0.765 | 1.443 | Peroxidase, MnP | 8.86 | 8.84 | 0.988 | 0.74100 | |
| 169968 | 0.816 | 0.718 | 0.369 | 0.032 | 42.96 | 1.275 | 1.785 | 0.359 | 1.434 | Peroxidase, MnP | 10.27 | 10.16 | 0.930 | 0.44200 | |
| Laccase | 84170 | 0.741 | 0.664 | 0.325 | 0.032 | 58.64 | −0.210 | 0.085 | 0.338 | −0.517 | laccase | 10.45 | 10.34 | 0.931 | 0.17000 |
| 88089 | 0.841 | 0.684 | 0.336 | 0.039 | 41.00 | 1.770 | 0.715 | 0.695 | −0.016 | Laccase | 9.38 | 9.29 | 0.939 | 0.17800 | |
| 120834 | 0.737 | 0.669 | 0.330 | 0.011 | 57.80 | −0.289 | 0.242 | −0.825 | −0.271 | Laccase | 10.74 | 10.66 | 0.945 | 0.50800 | |
| 127045 | 0.752 | 0.681 | 0.339 | 0.024 | 54.32 | 0.008 | 0.620 | −0.103 | 0.112 | Laccase | 10.54 | 10.02 | 0.694 | 0.00260 | |
| 127050 | 0.721 | 0.667 | 0.334 | 0.015 | 60.26 | −0.606 | 0.179 | −0.581 | −0.098 | Laccase | 11.35 | 11.19 | 0.895 | 0.54300 | |
| 130783 | 0.791 | 0.705 | 0.347 | 0.037 | 43.13 | 0.780 | 1.376 | 0.605 | 0.461 | Laccase | 11.02 | 13.77 | 6.766 | 0.00426 | |
| 149668 | 0.775 | 0.679 | 0.334 | 0.019 | 51.25 | 0.463 | 0.557 | −0.397 | −0.120 | Laccase | 9.91 | 9.81 | 0.931 | 0.35300 | |
| Cellulose Binding Protein | 59733 | 0.812 | 0.704 | 0.346 | 0.026 | 46.11 | 1.196 | 1.344 | −0.018 | 0.414 | GH10-CBM1 | 9.24 | 13.69 | 21.783 | 0.00004 |
| 66688 | 0.824 | 0.716 | 0.347 | 0.025 | 47.02 | 1.434 | 1.722 | −0.048 | 0.488 | GH61-CBM1 | 9.62 | 14.87 | 37.901 | 0.00002 | |
| 67561 | 0.853 | 0.730 | 0.366 | 0.020 | 39.43 | 2.008 | 2.163 | −0.336 | 1.313 | GH10-CBM1 | 10.30 | 13.71 | 10.642 | 0.00002 | |
| 68569 | 0.837 | 0.735 | 0.345 | 0.024 | 43.59 | 1.691 | 2.321 | −0.095 | 0.383 | CE1-CBM1 | 9.80 | 13.10 | 9.858 | 0.00023 | |
| 79557 | 0.802 | 0.715 | 0.345 | 0.022 | 42.15 | 0.998 | 1.691 | −0.231 | 0.373 | GH5-CBM1 | 10.24 | 14.04 | 13.874 | 0.00001 | |
| 87580 | 0.783 | 0.693 | 0.321 | 0.010 | 49.31 | 0.622 | 0.998 | −0.884 | −0.692 | CE16-CBM1 | 10.94 | 14.46 | 11.540 | 0.00004 | |
| 89533 | 0.838 | 0.736 | 0.353 | 0.052 | 42.06 | 1.711 | 2.352 | 1.426 | 0.719 | GH61-CBM1 | 10.35 | 13.56 | 9.306 | 0.00002 | |
| 89534 | 0.870 | 0.731 | 0.352 | 0.044 | 37.58 | 2.345 | 2.195 | 0.980 | 0.693 | GH61-CBM1 | 9.42 | 10.13 | 1.637 | 0.02300 | |
| 101925 | 0.845 | 0.723 | 0.359 | 0.033 | 38.22 | 1.850 | 1.943 | 0.394 | 1.020 | GH7-CBM1 | 8.84 | 8.96 | 1.086 | 0.09060 | |
| 106777 | 0.804 | 0.712 | 0.348 | 0.035 | 49.87 | 1.038 | 1.596 | 0.511 | 0.500 | GH5- CBM1 | 9.54 | 14.35 | 28.050 | 0.00001 | |
| 109840 | 0.857 | 0.741 | 0.370 | 0.039 | 38.16 | 2.087 | 2.509 | 0.733 | 1.471 | GH10- CBM1 | 9.52 | 11.85 | 5.009 | 0.00053 | |
| 129028 | 0.852 | 0.733 | 0.363 | 0.042 | 39.91 | 1.988 | 2.258 | 0.849 | 1.181 | GH5- CBM1 | 9.81 | 10.79 | 1.985 | 0.01830 | |
| 133809 | 0.865 | 0.713 | 0.338 | 0.040 | 37.08 | 2.246 | 1.628 | 0.779 | 0.091 | GH11-CBM1 | 10.86 | 11.42 | 1.466 | 0.02090 | |
| 148588 | 0.856 | 0.742 | 0.376 | 0.008 | 34.45 | 2.067 | 2.541 | −0.967 | 1.756 | GH7-CBM1 | 11.02 | 12.62 | 3.015 | 0.00018 | |
| CDH | 84792 | 0.803 | 0.688 | 0.338 | 0.023 | 48.09 | 1.018 | 0.841 | −0.137 | 0.071 | CDH | 9.29 | 13.76 | 22.241 | 0.00002 |
| 87110 | 0.769 | 0.679 | 0.332 | 0.024 | 50.71 | 0.345 | 0.557 | −0.111 | −0.189 | 11.31 | 11.04 | 0.827 | 0.20200 | ||
| 125610 | 0.762 | 0.665 | 0.318 | 0.044 | 53.34 | 0.206 | 0.116 | 0.981 | −0.814 | cir1 CBM1 | 10.17 | 10.40 | 1.170 | 0.23300 | |
| 147544 | 0.712 | 0.665 | 0.318 | 0.025 | 56.98 | −0.784 | 0.116 | −0.060 | −0.798 | 11.24 | 11.02 | 0.860 | 0.19900 | ||
| Delta 12 Dehidrogenase | 58880 | 0.727 | 0.670 | 0.317 | 0.008 | 59.40 | −0.487 | 0.274 | −0.973 | −0.853 | Δ-12 FAD | 10.36 | 10.29 | 0.956 | 0.72900 |
| 121074 | 0.731 | 0.644 | 0.313 | 0.022 | 55.49 | −0.408 | −0.545 | −0.219 | −1.033 | Δ-12 FAD | 10.58 | 10.23 | 0.783 | 0.01490 | |
| 124050 | 0.856 | 0.736 | 0.368 | 0.042 | 38.22 | 2.067 | 2.352 | 0.875 | 1.416 | Δ-12 FAD. Cs-fad2 | 12.74 | 12.66 | 0.941 | 0.65300 | |
| 136101 | 0.714 | 0.649 | 0.310 | 0.007 | 56.78 | −0.745 | −0.387 | −1.016 | −1.178 | Δ-12 FAD | 11.01 | 12.54 | 2.895 | 0.00022 | |
| 167690 | 0.736 | 0.670 | 0.325 | 0.013 | 58.45 | −0.309 | 0.274 | −0.706 | −0.484 | Δ-12 FAD | 10.67 | 10.11 | 0.678 | 0.00870 | |
| Delta 9 Dehidrogenase | 87875 | 0.810 | 0.694 | 0.343 | 0.035 | 44.18 | 1.156 | 1.030 | 0.475 | 0.313 | Δ-9 FAD, Cs-ole1 | 8.93 | 8.94 | 1.006 | 0.88000 |
| 129045 | 0.728 | 0.679 | 0.335 | 0.045 | 56.92 | −0.467 | 0.557 | 1.017 | −0.075 | Δ-9 FAD, Cs-ole1 | 8.95 | 8.91 | 0.974 | 0.52700 | |
| 129048 | 0.848 | 0.740 | 0.366 | 0.050 | 41.46 | 1.909 | 2.478 | 1.291 | 1.292 | Δ-9 FAD, Cs-ole1 | 11.78 | 12.35 | 1.478 | 0.02980 | |
| 133675 | 0.760 | 0.669 | 0.330 | 0.024 | 54.62 | 0.166 | 0.242 | −0.113 | −0.266 | Δ-9 FAD, Cs-ole1 | 9.64 | 9.51 | 0.917 | 0.17900 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gonzalez, A.; Corsini, G.; Lobos, S.; Seelenfreund, D.; Tello, M. Metabolic Specialization and Codon Preference of Lignocellulolytic Genes in the White Rot Basidiomycete Ceriporiopsis subvermispora. Genes 2020, 11, 1227. https://doi.org/10.3390/genes11101227
Gonzalez A, Corsini G, Lobos S, Seelenfreund D, Tello M. Metabolic Specialization and Codon Preference of Lignocellulolytic Genes in the White Rot Basidiomycete Ceriporiopsis subvermispora. Genes. 2020; 11(10):1227. https://doi.org/10.3390/genes11101227
Chicago/Turabian StyleGonzalez, Alex, Gino Corsini, Sergio Lobos, Daniela Seelenfreund, and Mario Tello. 2020. "Metabolic Specialization and Codon Preference of Lignocellulolytic Genes in the White Rot Basidiomycete Ceriporiopsis subvermispora" Genes 11, no. 10: 1227. https://doi.org/10.3390/genes11101227