Fine-Tuning of Alanyl-tRNA Synthetase Quality Control Alleviates Global Dysregulation of the Proteome

Abstract
:1. Introduction
2. Materials and Methods
2.1. General Methods
2.2. Suppressor Mutant Characterization
2.3. Strain Construction
2.4. Growth Analysis
2.5. Motility Assays
2.6. Antibiotic Sensitivity
2.7. In vivo Mistranslation Reporter
2.8. Preparation of Recombinant Protein and In Vitro Transcribed tRNAAla
2.9. Proofreading Activity
2.10. Pyrophosphate Exchange
3. Results
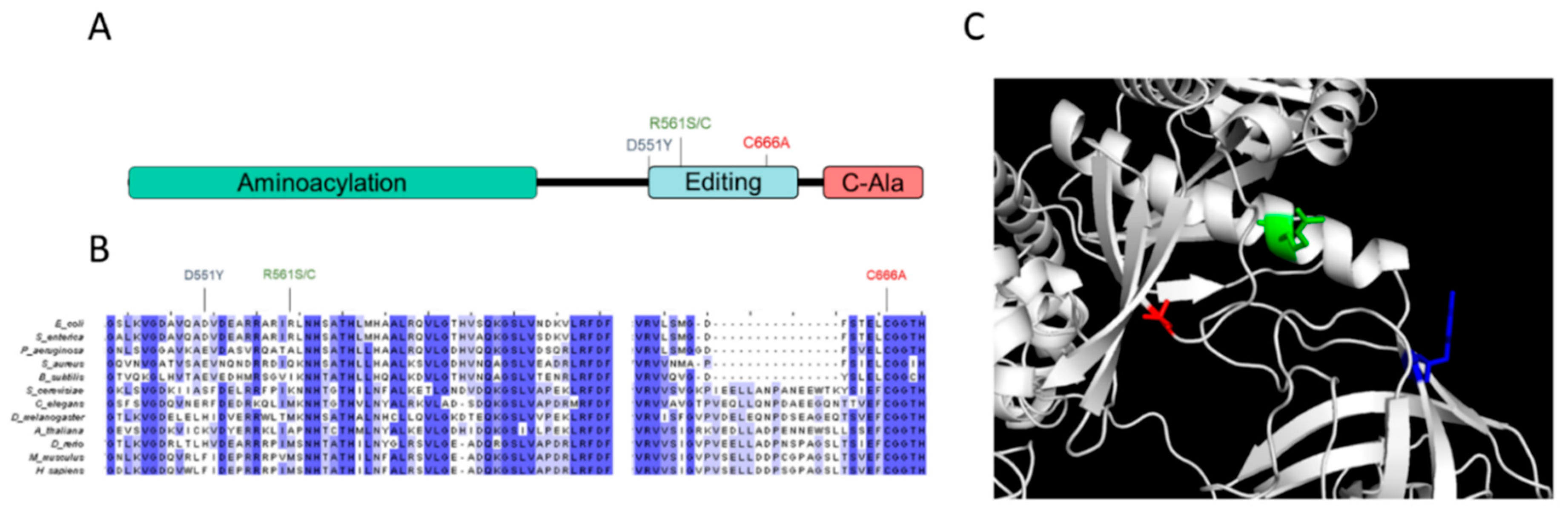
3.1. Identification of Suppressor Mutations That Alleviate the AlaRS C666A Growth Defect
3.2. Second-Site AlaRS Mutations Alleviate the Slow-Growth AlaRS C666A Phenotype
3.3. AlaRS C666A Suppressors Do Not Prevent Serine Mistranslation
3.4. Phenotypes Associated with AlaRS-Mediated Proteome Dysregulation Are Alleviated in with the Second-Site Suppressor Mutations
3.5. In Vitro Characterization of the AlaRS C666A Suppressors
3.6. AlaRS R561C Reduces Ser-tRNAAla Formation
3.7. AlaRS R561S Has Elevated Proofreading Activity
3.8. AlaRS D551Y Increases Ser-tRNAAla Deacylation in Trans
4. Discussion
4.1. Fitness Costs Associated with Translational Errors and Disruption of the Aminoacyl-tRNA Pool Are Amino Acid-Specific
4.2. Perturbation of AaRS Fidelity Leads to Rapid Acquisition of the AlaRS C666A Variant
4.3. Secondary AaRS Proofreading Activities Permit Regulation of Translational Quality Control
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lichtarge, O.; Bourne, H.R.; Cohen, F.E. An evolutionary trace method defines binding surfaces common to protein families. J. Mol. Biol. 1996, 257, 342–358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tondnevis, F.; Dudenhausen, E.E.; Miller, A.M.; McKenna, R.; Altschul, S.F.; Bloom, L.B.; Neuwald, A.F. Deep Analysis of Residue Constraints (DARC): Identifying determinants of protein functional specificity. Sci. Rep. 2020, 10, 1691. [Google Scholar] [CrossRef]
- Mohler, K.; Ibba, M. Translational fidelity and mistranslation in the cellular response to stress. Nat. Microbiol. 2017, 2, 17117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ibba, M.; Soll, D. Aminoacyl-tRNA synthesis. Annu. Rev. Biochem. 2000, 69, 617–650. [Google Scholar] [CrossRef] [PubMed]
- Yadavalli, S.S.; Ibba, M. Quality control in aminoacyl-tRNA synthesis its role in translational fidelity. Adv. Protein Chem. Struct. Biol. 2012, 86, 1–43. [Google Scholar] [CrossRef]
- Baldwin, A.N.; Berg, P. Transfer ribonucleic acid-induced hydrolysis of valyladenylate bound to isoleucyl ribonucleic acid synthetase. J. Biol. Chem. 1966, 241, 839–845. [Google Scholar]
- Roy, H.; Ling, J.; Irnov, M.; Ibba, M. Post-transfer editing in vitro and in vivo by the β subunit of phenylalanyl-tRNA synthetase. EMBO J. 2004, 23, 4639–4648. [Google Scholar] [CrossRef] [Green Version]
- Dock-Bregeon, A.C.; Rees, B.; Torres-Larios, A.; Bey, G.; Caillet, J.; Moras, D. Achieving error-free translation; the mechanism of proofreading of threonyl-tRNA synthetase at atomic resolution. Mol. Cell 2004, 16, 375–386. [Google Scholar] [CrossRef]
- Beebe, K.; Ribas De Pouplana, L.; Schimmel, P. Elucidation of tRNA-dependent editing by a class II tRNA synthetase and significance for cell viability. EMBO J. 2003, 22, 668–675. [Google Scholar] [CrossRef] [Green Version]
- Bullwinkle, T.J.; Reynolds, N.M.; Raina, M.; Moghal, A.; Matsa, E.; Rajkovic, A.; Kayadibi, H.; Fazlollahi, F.; Ryan, C.; Howitz, N.; et al. Oxidation of cellular amino acid pools leads to cytotoxic mistranslation of the genetic code. Elife 2014, 3, e02501. [Google Scholar] [CrossRef] [PubMed]
- Bullwinkle, T.J.; Ibba, M. Translation quality control is critical for bacterial responses to amino acid stress. Proc. Natl. Acad. Sci. USA 2016, 113, 2252–2257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohler, K.; Mann, R.; Bullwinkle, T.J.; Hopkins, K.; Hwang, L.; Reynolds, N.M.; Gassaway, B.; Aerni, H.R.; Rinehart, J.; Polymenis, M.; et al. Editing of misaminoacylated tRNA controls the sensitivity of amino acid stress responses in Saccharomyces cerevisiae. Nucleic Acids Res. 2017, 45, 3985–3996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelly, P.; Backes, N.; Mohler, K.; Buser, C.; Kavoor, A.; Rinehart, J.; Phillips, G.; Ibba, M. Alanyl-tRNA Synthetase Quality Control Prevents Global Dysregulation of the Escherichia coli Proteome. Mbio 2019, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Afgan, E.; Baker, D.; Batut, B.; van den Beek, M.; Bouvier, D.; Cech, M.; Chilton, J.; Clements, D.; Coraor, N.; Gruning, B.A.; et al. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2018 update. Nucleic Acids Res. 2018, 46, W537–W544. [Google Scholar] [CrossRef] [Green Version]
- Madeira, F.; Park, Y.M.; Lee, J.; Buso, N.; Gur, T.; Madhusoodanan, N.; Basutkar, P.; Tivey, A.R.N.; Potter, S.C.; Finn, R.D.; et al. The EMBL-EBI search and sequence analysis tools APIs in 2019. Nucleic Acids Res. 2019, 47, W636–W641. [Google Scholar] [CrossRef] [Green Version]
- Waterhouse, A.M.; Procter, J.B.; Martin, D.M.; Clamp, M.; Barton, G.J. Jalview Version 2--a multiple sequence alignment editor and analysis workbench. Bioinformatics 2009, 25, 1189–1191. [Google Scholar] [CrossRef] [Green Version]
- Kelley, L.A.; Mezulis, S.; Yates, C.M.; Wass, M.N.; Sternberg, M.J. The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 2015, 10, 845–858. [Google Scholar] [CrossRef] [Green Version]
- Reisch, C.R.; Prather, K.L. The no-SCAR (Scarless Cas9 Assisted Recombineering) system for genome editing in Escherichia coli. Sci. Rep. 2015, 5, 15096. [Google Scholar] [CrossRef] [Green Version]
- Reisch, C.R.; Prather, K.L.J. Scarless Cas9 Assisted Recombineering (no-SCAR) in Escherichia coli, an Easy-to-Use System for Genome Editing. Curr. Protoc. Mol. Biol. 2017, 117, 31–38. [Google Scholar] [CrossRef]
- Wilkinson, A.J.; Fersht, A.R.; Blow, D.M.; Winter, G. Site-directed mutagenesis as a probe of enzyme structure and catalysis: Tyrosyl-tRNA synthetase cysteine-35 to glycine-35 mutation. Biochemistry 1983, 22, 3581–3586. [Google Scholar] [CrossRef]
- Steiner, R.E.; Kyle, A.M.; Ibba, M. Oxidation of phenylalanyl-tRNA synthetase positively regulates translational quality control. Proc. Natl. Acad. Sci. USA 2019, 116, 10058–10063. [Google Scholar] [CrossRef] [Green Version]
- LaRiviere, F.J.; Wolfson, A.D.; Uhlenbeck, O.C. Uniform binding of aminoacyl-tRNAs to elongation factor Tu by thermodynamic compensation. Science 2001, 294, 165–168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanderson, L.E.; Uhlenbeck, O.C. Directed mutagenesis identifies amino acid residues involved in elongation factor Tu binding to yeast Phe-tRNAPhe. J. Mol. Biol. 2007, 368, 119–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanderson, L.E.; Uhlenbeck, O.C. Exploring the specificity of bacterial elongation factor Tu for different tRNAs. Biochemistry 2007, 46, 6194–6200. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Vargas-Rodriguez, O.; Goto, Y.; Novoa, E.M.; Ribas de Pouplana, L.; Suga, H.; Musier-Forsyth, K. Homologous trans-editing factors with broad tRNA specificity prevent mistranslation caused by serine/threonine misactivation. Proc. Natl. Acad. Sci. USA 2015, 112, 6027–6032. [Google Scholar] [CrossRef] [Green Version]
- Shi, J.P.; Musier-Forsyth, K.; Schimmel, P. Region of a conserved sequence motif in a class II tRNA synthetase needed for transfer of an activated amino acid to an RNA substrate. Biochemistry 1994, 33, 5312–5318. [Google Scholar] [CrossRef]
- Guo, M.; Chong, Y.E.; Shapiro, R.; Beebe, K.; Yang, X.L.; Schimmel, P. Paradox of mistranslation of serine for alanine caused by AlaRS recognition dilemma. Nature 2009, 462, 808–812. [Google Scholar] [CrossRef] [Green Version]
- Jasin, M.; Regan, L.; Schimmel, P. Dispensable pieces of an aminoacyl tRNA synthetase which activate the catalytic site. Cell 1984, 36, 1089–1095. [Google Scholar] [CrossRef]
- Guo, M.; Chong, Y.E.; Beebe, K.; Shapiro, R.; Yang, X.L.; Schimmel, P. The C-Ala domain brings together editing and aminoacylation functions on one tRNA. Science 2009, 325, 744–747. [Google Scholar] [CrossRef] [Green Version]
- Pasman, Z.; Robey-Bond, S.; Mirando, A.C.; Smith, G.J.; Lague, A.; Francklyn, C.S. Substrate specificity and catalysis by the editing active site of Alanyl-tRNA synthetase from Escherichia coli. Biochemistry 2011, 50, 1474–1482. [Google Scholar] [CrossRef] [Green Version]
- Ruan, B.; Palioura, S.; Sabina, J.; Marvin-Guy, L.; Kochhar, S.; Larossa, R.A.; Soll, D. Quality control despite mistranslation caused by an ambiguous genetic code. Proc. Natl. Acad. Sci. USA 2008, 105, 16502–16507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haseltine, W.A.; Block, R. Synthesis of guanosine tetra- and pentaphosphate requires the presence of a codon-specific, uncharged transfer ribonucleic acid in the acceptor site of ribosomes. Proc. Natl. Acad. Sci. USA 1973, 70, 1564–1568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hauryliuk, V.; Atkinson, G.C.; Murakami, K.S.; Tenson, T.; Gerdes, K. Recent functional insights into the role of (p) ppGpp in bacterial physiology. Nat. Rev. Microbiol. 2015, 13, 298–309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stent, G.S.; Brenner, S. A genetic locus for the regulation of ribonucleic acid synthesis. Proc. Natl. Acad. Sci. USA 1961, 47, 2005–2014. [Google Scholar] [CrossRef] [Green Version]
- Bacher, J.M.; Schimmel, P. An editing-defective aminoacyl-tRNA synthetase is mutagenic in aging bacteria via the SOS response. Proc. Natl. Acad. Sci. USA 2007, 104, 1907–1912. [Google Scholar] [CrossRef] [Green Version]
- Samhita, L.; Raval, P.K.; Agashe, D. Global mistranslation increases cell survival under stress in Escherichia coli. PLoS Genet. 2020, 16, e1008654. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.W.; Beebe, K.; Nangle, L.A.; Jang, J.; Longo-Guess, C.M.; Cook, S.A.; Davisson, M.T.; Sundberg, J.P.; Schimmel, P.; Ackerman, S.L. Editing-defective tRNA synthetase causes protein misfolding and neurodegeneration. Nature 2006, 443, 50–55. [Google Scholar] [CrossRef]
- Liu, Y.; Satz, J.S.; Vo, M.N.; Nangle, L.A.; Schimmel, P.; Ackerman, S.L. Deficiencies in tRNA synthetase editing activity cause cardioproteinopathy. Proc. Natl. Acad. Sci. USA 2014, 111, 17570–17575. [Google Scholar] [CrossRef] [Green Version]
- Hilander, T.; Zhou, X.L.; Konovalova, S.; Zhang, F.P.; Euro, L.; Chilov, D.; Poutanen, M.; Chihade, J.; Wang, E.D.; Tyynismaa, H. Editing activity for eliminating mischarged tRNAs is essential in mammalian mitochondria. Nucleic Acids Res. 2018, 46, 849–860. [Google Scholar] [CrossRef] [Green Version]
- Cvetesic, N.; Perona, J.J.; Gruic-Sovulj, I. Kinetic partitioning between synthetic and editing pathways in class I aminoacyl-tRNA synthetases occurs at both pre-transfer and post-transfer hydrolytic steps. J. Biol. Chem. 2012, 287, 25381–25394. [Google Scholar] [CrossRef] [Green Version]
- Cvetesic, N.; Palencia, A.; Halasz, I.; Cusack, S.; Gruic-Sovulj, I. The physiological target for LeuRS translational quality control is norvaline. EMBO J. 2014, 33, 1639–1653. [Google Scholar] [CrossRef] [PubMed]
- Ling, J.; Soll, D. Severe oxidative stress induces protein mistranslation through impairment of an aminoacyl-tRNA synthetase editing site. Proc. Natl. Acad. Sci. USA 2010, 107, 4028–4033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.; Venkat, S.; Hudson, D.; Wang, T.; Gan, Q.; Fan, C. Site-Specifically Studying Lysine Acetylation of Aminoacyl-tRNA Synthetases. ACS Chem. Biol. 2019, 14, 288–295. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ala | Ser a (kcat/Km) | Specificity b (Ala/Ser) | |||
|---|---|---|---|---|---|
| Km | kcat | kcat/Km | |||
| µM | min−1 | min−1/µM | min−1/µM | ||
| E. coli AlaRS | 320 ± 150 | 1246 ± 105 | 3.91 | 4.7 × 10−3 ± 1.5 × 10−4 | 832 |
| AlaRS C666A | 263 ± 115 | 1010 ± 380 | 3.83 | 7.3 × 10−3 ± 5 × 10−4 | 525 |
| AlaRS D551Y | 260 ± 76 | 715 ± 220 | 2.76 | 6.9 × 10−3 ± 2.5 × 10−4 | 400 |
| AlaRS D551Y C666A | 217 ± 100 | 1096 ± 265 | 5.06 | 10.4 × 10−3 ± 14 × 10−4 | 490 |
| AlaRS R561C | 203 ± 68 | 1306 ± 345 | 6.45 | 6.7 × 10−3 ± 13 × 10−4 | 963 |
| AlaRS R561C C666A | 190 ± 30 | 819 ± 88 | 4.29 | 5.5 × 10−3 ± 19 × 10−4 | 780 |
| AlaRS R561S | 180 ± 15 | 1216 ± 125 | 6.70 | 8.2 × 10−3 ± 19 × 10−4 | 817 |
| AlaRS R561S C666A | 177 ± 37 | 791 ± 150 | 4.46 | 7.3 × 10−3 ± 5 × 10−4 | 611 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kelly, P.; Kavoor, A.; Ibba, M. Fine-Tuning of Alanyl-tRNA Synthetase Quality Control Alleviates Global Dysregulation of the Proteome. Genes 2020, 11, 1222. https://doi.org/10.3390/genes11101222
Kelly P, Kavoor A, Ibba M. Fine-Tuning of Alanyl-tRNA Synthetase Quality Control Alleviates Global Dysregulation of the Proteome. Genes. 2020; 11(10):1222. https://doi.org/10.3390/genes11101222
Chicago/Turabian StyleKelly, Paul, Arundhati Kavoor, and Michael Ibba. 2020. "Fine-Tuning of Alanyl-tRNA Synthetase Quality Control Alleviates Global Dysregulation of the Proteome" Genes 11, no. 10: 1222. https://doi.org/10.3390/genes11101222