Hepatic Transcriptomics Reveals that Lipogenesis Is a Key Signaling Pathway in Isocitrate Dehydrogenase 2 Deficient Mice

 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
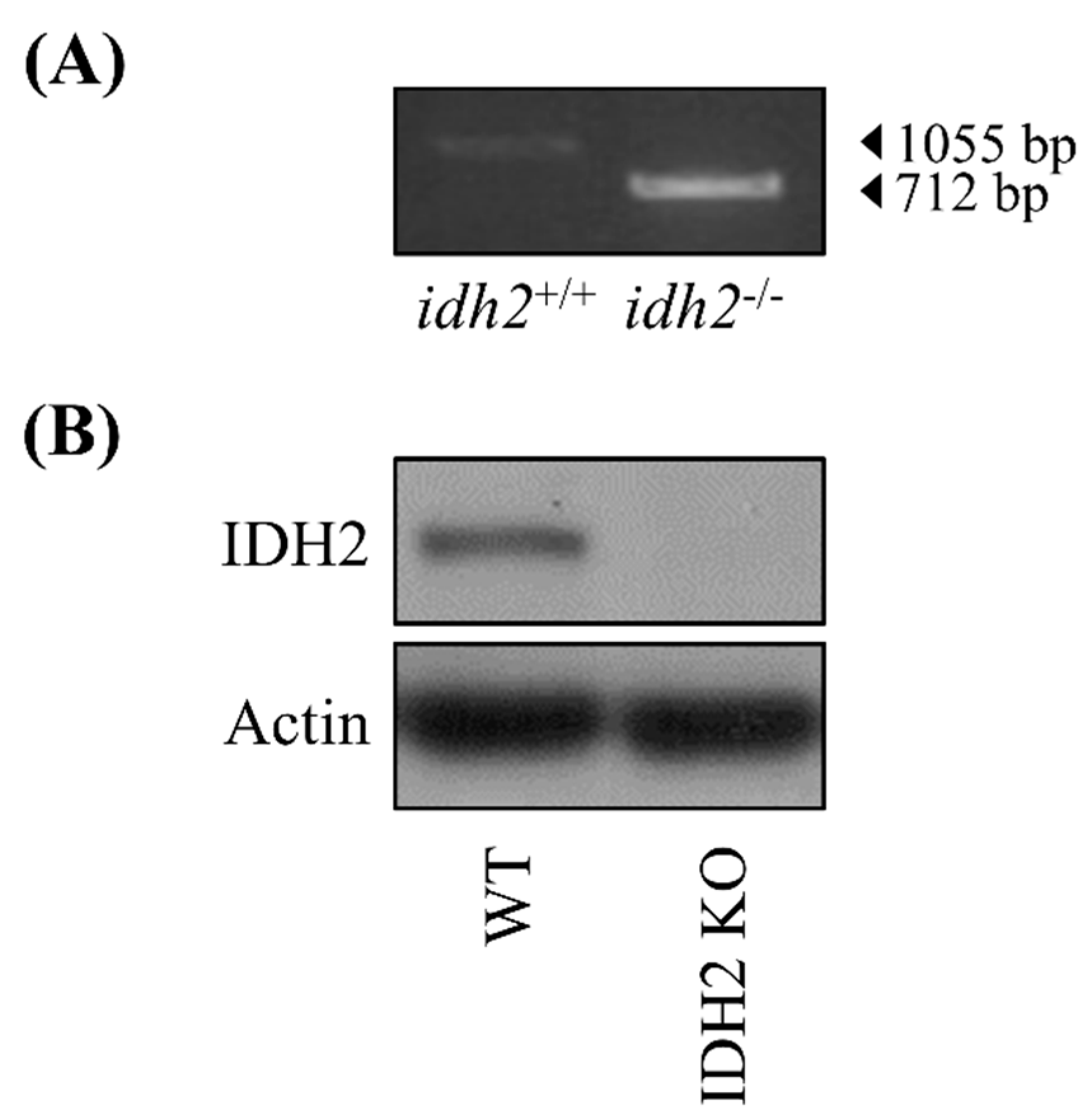
2.1. Animal Housing, and Study Design
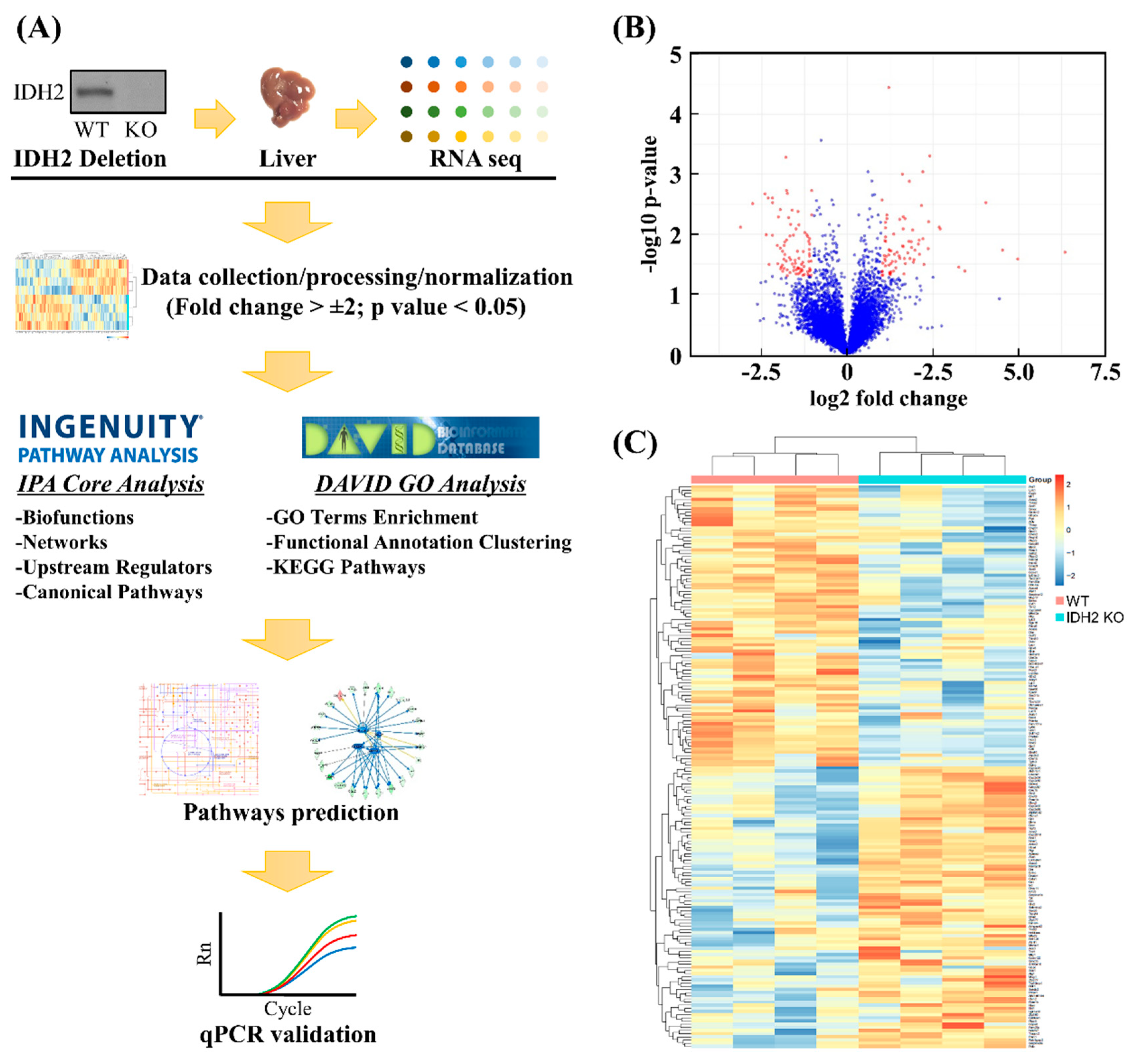
2.2. Liver Transcriptomics Analysis
2.3. Bioinformatics Analyses
2.4. Quantitative RT-PCR Validation
2.5. Statistical Analyses
3. Results
3.1. Transcriptome Profiles and Identification of DEGs
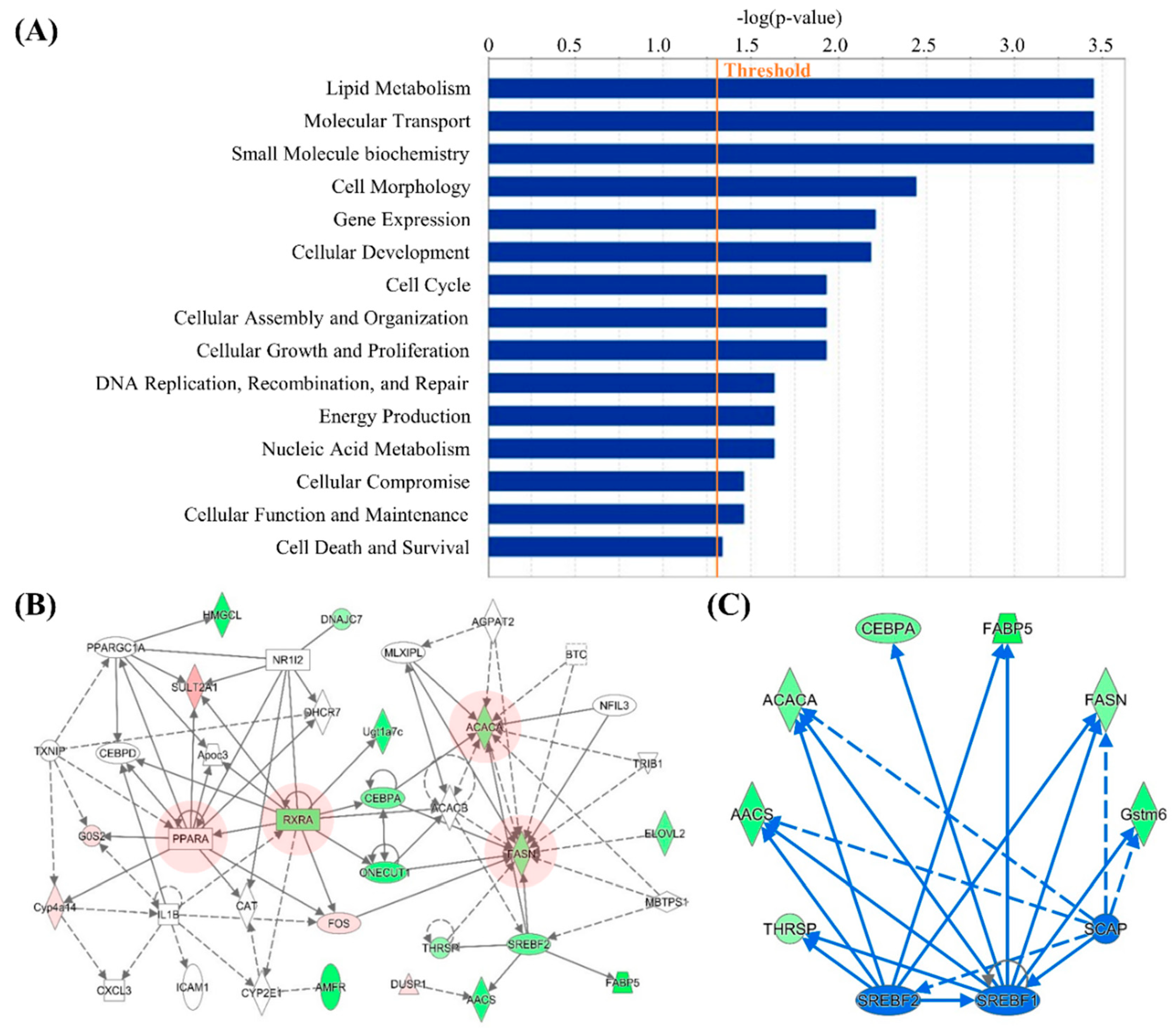
3.2. Computational Bioinformatic Pathway Prediction of DEGs
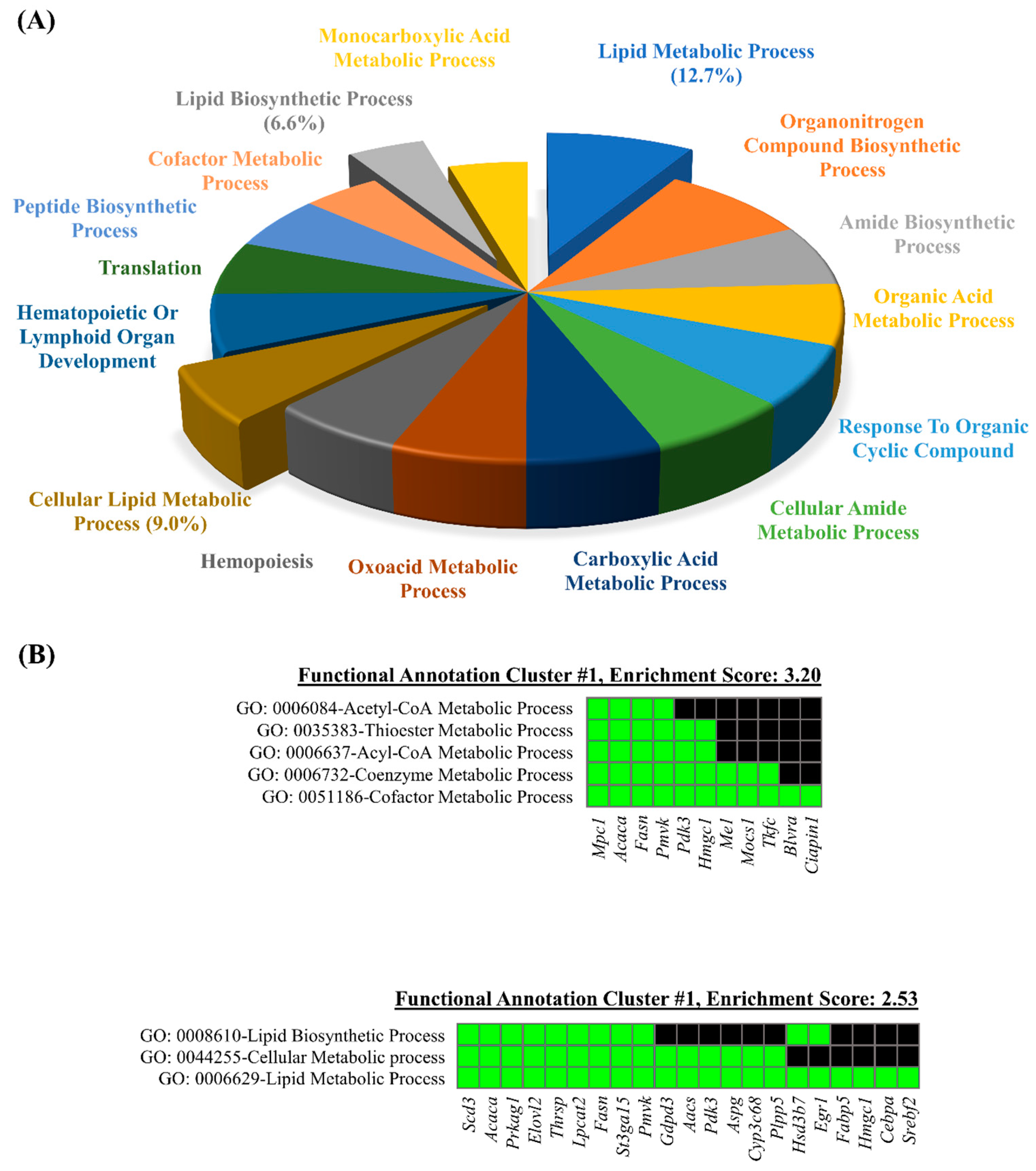
3.3. Database for Annotation, Visualization, and Integrated Discovery (DAVID) Web-Based Bioinformatic Analysis of DEGs
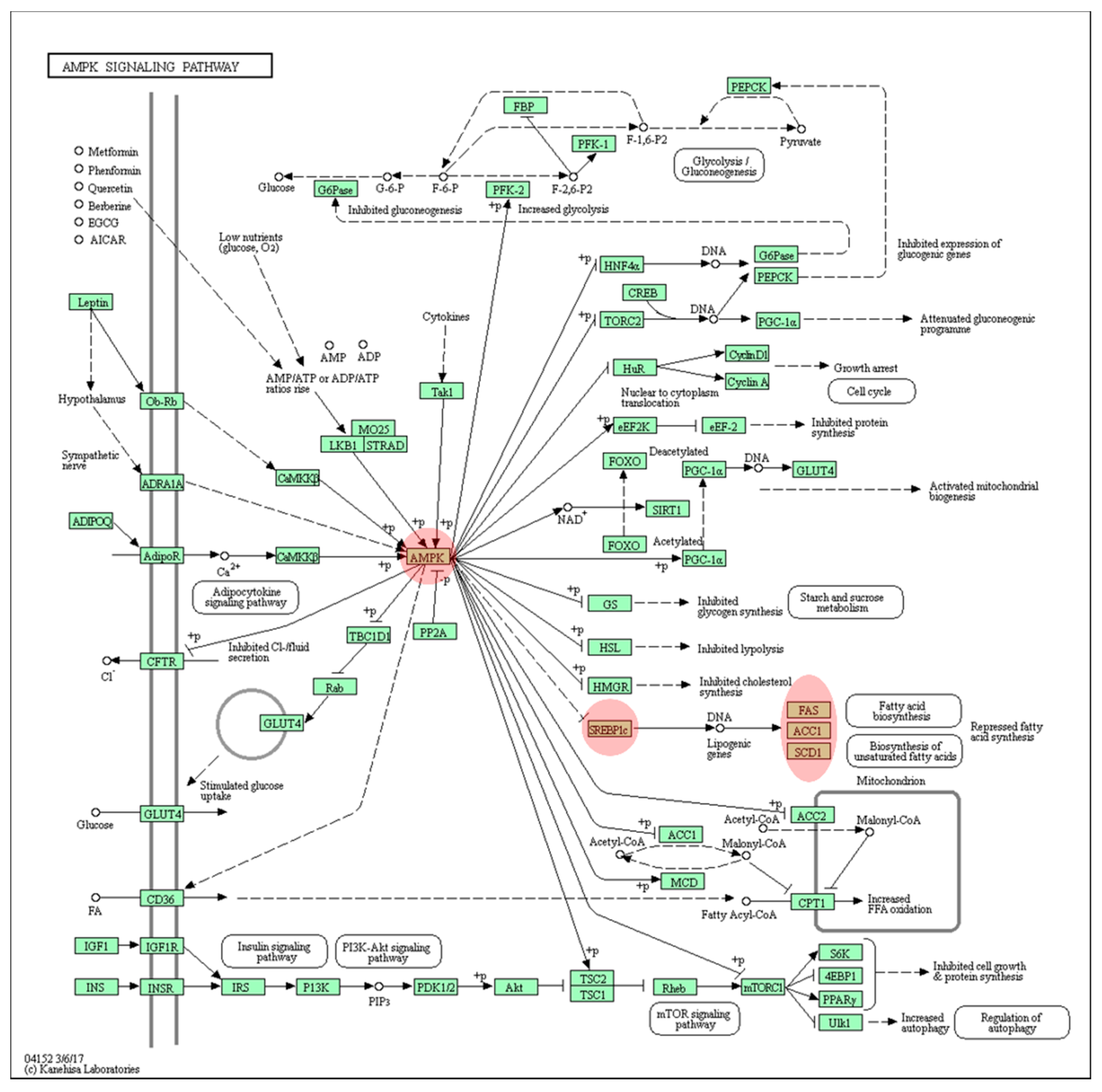
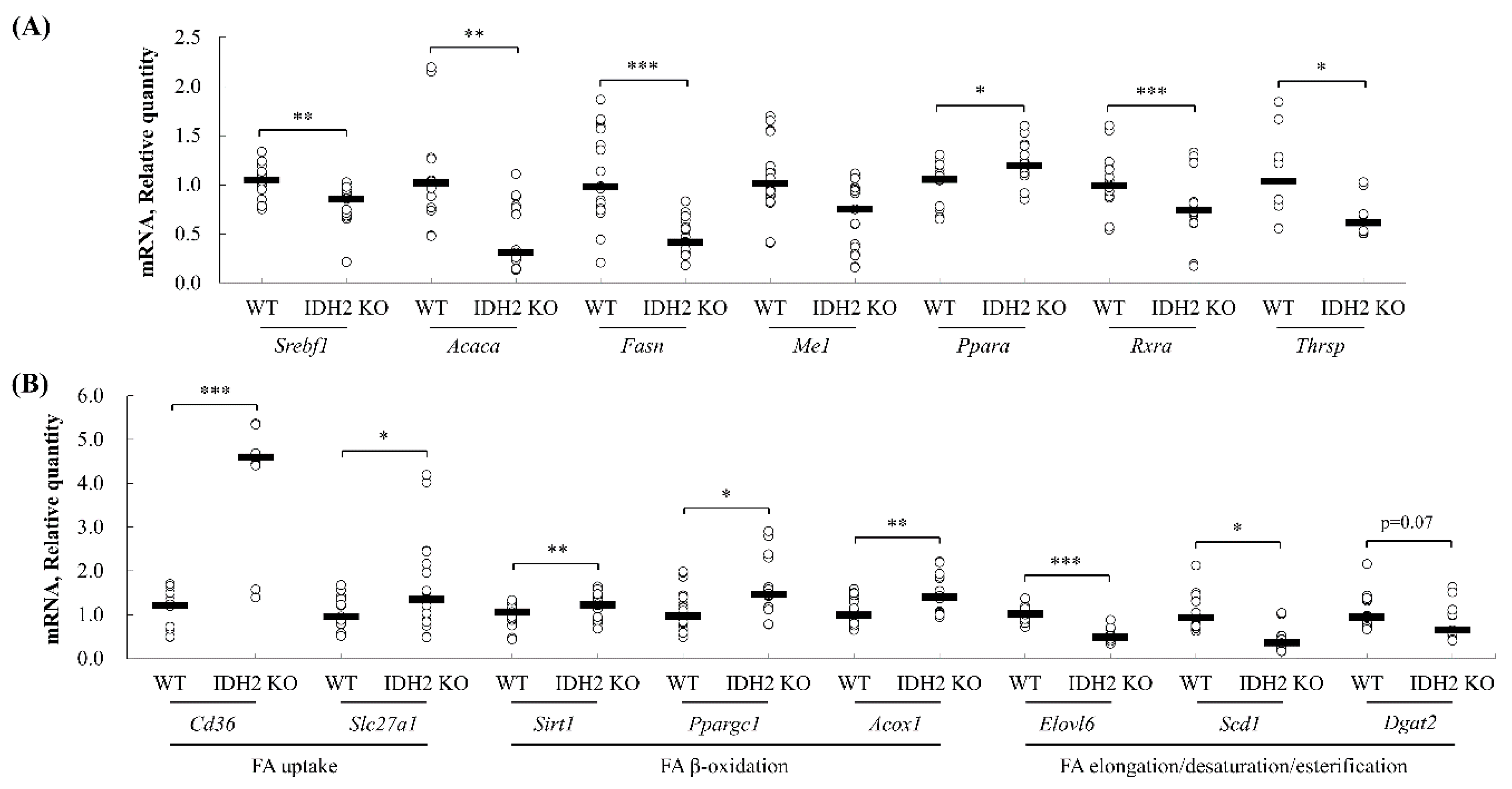
3.4. Quantitative PCR Validation of Predicted Pathway of DEGs
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Jo, S.H.; Son, M.K.; Koh, H.J.; Lee, S.M.; Song, I.H.; Kim, Y.O.; Lee, Y.S.; Jeong, K.S.; Kim, W.B.; Park, J.W.; et al. Control of mitochondrial redox balance and cellular defense against oxidative damage by mitochondrial NADP+-dependent isocitrate dehydrogenase. J. Biol. Chem. 2001, 276, 16168–16176. [Google Scholar] [CrossRef] [PubMed]
- Koshland, D.E., Jr.; Walsh, K.; LaPorte, D.C. Sensitivity of metabolic fluxes to covalent control. Curr. Top. Cell. Regul. 1985, 27, 13–22. [Google Scholar] [PubMed]
- Reitman, Z.J.; Yan, H. Isocitrate dehydrogenase 1 and 2 mutations in cancer: Alterations at a crossroads of cellular metabolism. J. Natl. Cancer Inst. 2010, 102, 932–941. [Google Scholar] [CrossRef] [PubMed]
- Veech, R.L.; Eggleston, L.V.; Krebs, H.A. The redox state of free nicotinamide-adenine dinucleotide phosphate in the cytoplasm of rat liver. Biochem. J. 1969, 115, 609–619. [Google Scholar] [CrossRef] [PubMed]
- Berg, J.; Tymoczko, J.; Stryer, L. Section 22.4 fatty acids are synthesized and degraded by different pathways. In Biochemistry, 5th ed.; W. H. Freeman and Company: New York, NY, USA, 2002; p. 2013. [Google Scholar]
- Cha, H.; Lee, S.; Hwan Kim, S.; Kim, H.; Lee, D.S.; Lee, H.S.; Lee, J.H.; Park, J.W. Increased susceptibility of IDH2-deficient mice to dextran sodium sulfate-induced colitis. Redox Biol. 2017, 13, 32–38. [Google Scholar] [CrossRef]
- Ku, H.J.; Ahn, Y.; Lee, J.H.; Park, K.M.; Park, J.W. IDH2 deficiency promotes mitochondrial dysfunction and cardiac hypertrophy in mice. Free Radic. Biol. Med. 2015, 80, 84–92. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.J.; Kim, S.H.; Park, K.M.; Lee, J.H.; Park, J.W. Increased obesity resistance and insulin sensitivity in mice lacking the isocitrate dehydrogenase 2 gene. Free Radic. Biol. Med. 2016, 99, 179–188. [Google Scholar] [CrossRef]
- Han, S.J.; Choi, H.S.; Kim, J.I.; Park, J.W.; Park, K.M. IDH2 deficiency increases the liver susceptibility to ischemia-reperfusion injury via increased mitochondrial oxidative injury. Redox Biol. 2018, 14, 142–153. [Google Scholar] [CrossRef]
- Kong, M.J.; Han, S.J.; Kim, J.I.; Park, J.W.; Park, K.M. Mitochondrial NADP(+)-dependent isocitrate dehydrogenase deficiency increases cisplatin-induced oxidative damage in the kidney tubule cells. Cell Death Dis. 2018, 9, 488. [Google Scholar] [CrossRef]
- Kil, I.S.; Park, J.W. Regulation of mitochondrial NADP+-dependent isocitrate dehydrogenase activity by glutathionylation. J. Biol. Chem. 2005, 280, 10846–10854. [Google Scholar] [CrossRef]
- Kim, H.; Pan, J.H.; Kim, S.H.; Lee, J.H.; Park, J.W. Chlorogenic acid ameliorates alcohol-induced liver injuries through scavenging reactive oxygen species. Biochimie 2018, 150, 131–138. [Google Scholar] [CrossRef]
- Someya, S.; Yu, W.; Hallows, W.C.; Xu, J.; Vann, J.M.; Leeuwenburgh, C.; Tanokura, M.; Denu, J.M.; Prolla, T.A. Sirt3 mediates reduction of oxidative damage and prevention of age-related hearing loss under caloric restriction. Cell 2010, 143, 802–812. [Google Scholar] [CrossRef]
- Yu, W.; Dittenhafer-Reed, K.E.; Denu, J.M. SIRT3 protein deacetylates isocitrate dehydrogenase 2 (IDH2) and regulates mitochondrial redox status. J. Biol. Chem. 2012, 287, 14078–14086. [Google Scholar] [CrossRef]
- Mortazavi, A.; Williams, B.A.; McCue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 2008, 5, 621–628. [Google Scholar] [CrossRef]
- Kong, B.W.; Hudson, N.; Seo, D.; Lee, S.; Khatri, B.; Lassiter, K.; Cook, D.; Piekarski, A.; Dridi, S.; Anthony, N.; et al. RNA sequencing for global gene expression associated with muscle growth in a single male modern broiler line compared to a foundational Barred Plymouth Rock chicken line. BMC Genom. 2017, 18, 82. [Google Scholar] [CrossRef]
- Pan, J.H.; Kim, H.S.; Beane, K.E.; Montalbano, A.M.; Lee, J.H.; Kim, Y.J.; Kim, J.H.; Kong, B.C.; Kim, S.; Park, J.W.; et al. IDH2 Deficiency Aggravates Fructose-Induced NAFLD by Modulating Hepatic Fatty Acid Metabolism and Activating Inflammatory Signaling in Female Mice. Nutrients 2018, 10, 679. [Google Scholar] [CrossRef]
- Duntas, L.H.; Brenta, G. A renewed focus on the association between thyroid hormones and lipid metabolism. Front. Endocrinol. 2018, 9, 511. [Google Scholar] [CrossRef]
- Liu, Y.Y.; Brent, G.A. Thyroid hormone crosstalk with nuclear receptor signaling in metabolic regulation. Trends Endocrinol. Metab. 2010, 21, 166–173. [Google Scholar] [CrossRef] [Green Version]
- Koenig, R.J. Thyroid hormone receptor coactivators and corepressors. Thyroid 1998, 8, 703–713. [Google Scholar] [CrossRef]
- Brownsey, R.W.; Boone, A.N.; Elliott, J.E.; Kulpa, J.E.; Lee, W.M. Regulation of acetyl-CoA carboxylase. Biochem. Soc. Trans. 2006, 34, 223–227. [Google Scholar] [CrossRef]
- Mao, J.; DeMayo, F.J.; Li, H.; Abu-Elheiga, L.; Gu, Z.; Shaikenov, T.E.; Kordari, P.; Chirala, S.S.; Heird, W.C.; Wakil, S.J. Liver-specific deletion of acetyl-CoA carboxylase 1 reduces hepatic triglyceride accumulation without affecting glucose homeostasis. Proc. Natl. Acad. Sci. USA 2006, 103, 8552–8557. [Google Scholar] [CrossRef] [Green Version]
- Strable, M.S.; Ntambi, J.M. Genetic control of de novo lipogenesis: Role in diet-induced obesity. Crit. Rev. Biochem. Mol. Biol. 2010, 45, 199–214. [Google Scholar] [CrossRef]
- Lee, S.J.; Cha, H.; Kim, H.; Lee, J.H.; Park, J.W. Amelioration of late-onset hepatic steatosis in IDH2-deficient mice. Free Radic. Res. 2017, 51, 368–374. [Google Scholar] [CrossRef]
- Horton, J.D.; Bashmakov, Y.; Shimomura, I.; Shimano, H. Regulation of sterol regulatory element binding proteins in livers of fasted and refed mice. Proc. Natl. Acad. Sci. USA 1998, 95, 5987–5992. [Google Scholar] [CrossRef] [Green Version]
- Horton, J.D.; Goldstein, J.L.; Brown, M.S. SREBPs: Activators of the complete program of cholesterol and fatty acid synthesis in the liver. J. Clin. Investig. 2002, 109, 1125–1131. [Google Scholar] [CrossRef]
- Horton, J.D.; Shimomura, I.; Brown, M.S.; Hammer, R.E.; Goldstein, J.L.; Shimano, H. Activation of cholesterol synthesis in preference to fatty acid synthesis in liver and adipose tissue of transgenic mice overproducing sterol regulatory element-binding protein-2. J. Clin. Investig. 1998, 101, 2331–2339. [Google Scholar] [CrossRef]
- Shimomura, I.; Shimano, H.; Korn, B.S.; Bashmakov, Y.; Horton, J.D. Nuclear sterol regulatory element-binding proteins activate genes responsible for the entire program of unsaturated fatty acid biosynthesis in transgenic mouse liver. J. Biol. Chem. 1998, 273, 35299–35306. [Google Scholar] [CrossRef]
- Ji, C.; Kaplowitz, N. Betaine decreases hyperhomocysteinemia, endoplasmic reticulum stress, and liver injury in alcohol-fed mice. Gastroenterology 2003, 124, 1488–1499. [Google Scholar] [CrossRef]
- Wu, J.; Wang, C.; Li, S.; Li, S.; Wang, W.; Li, J.; Chi, Y.; Yang, H.; Kong, X.; Zhou, Y.; et al. Thyroid hormone-responsive SPOT 14 homolog promotes hepatic lipogenesis, and its expression is regulated by liver X receptor alpha through a sterol regulatory element-binding protein 1c-dependent mechanism in mice. Hepatology 2013, 58, 617–628. [Google Scholar] [CrossRef]
- You, M.; Fischer, M.; Deeg, M.A.; Crabb, D.W. Ethanol induces fatty acid synthesis pathways by activation of sterol regulatory element-binding protein (SREBP). J. Biol. Chem. 2002, 277, 29342–29347. [Google Scholar] [CrossRef]
- Yin, L.; Zhang, Y.; Hillgartner, F.B. Sterol regulatory element-binding protein-1 interacts with the nuclear thyroid hormone receptor to enhance acetyl-CoA carboxylase-alpha transcription in hepatocytes. J. Biol. Chem. 2002, 277, 19554–19565. [Google Scholar] [CrossRef] [PubMed]
- Amemiya-Kudo, M.; Shimano, H.; Hasty, A.H.; Yahagi, N.; Yoshikawa, T.; Matsuzaka, T.; Okazaki, H.; Tamura, Y.; Iizuka, Y.; Ohashi, K.; et al. Transcriptional activities of nuclear SREBP-1a, -1c, and -2 to different target promoters of lipogenic and cholesterogenic genes. J. Lipid Res. 2002, 43, 1220–1235. [Google Scholar] [PubMed]
- Shechter, I.; Dai, P.; Huo, L.; Guan, G. IDH1 gene transcription is sterol regulated and activated by SREBP-1a and SREBP-2 in human hepatoma HepG2 cells: Evidence that IDH1 may regulate lipogenesis in hepatic cells. J. Lipid Res. 2003, 44, 2169–2180. [Google Scholar] [CrossRef]
- Carling, D.; Zammit, V.A.; Hardie, D.G. A common bicyclic protein kinase cascade inactivates the regulatory enzymes of fatty acid and cholesterol biosynthesis. FEBS Lett. 1987, 223, 217–222. [Google Scholar] [CrossRef] [Green Version]
- Oakhill, J.S.; Steel, R.; Chen, Z.P.; Scott, J.W.; Ling, N.; Tam, S.; Kemp, B.E. AMPK is a direct adenylate charge-regulated protein kinase. Science 2011, 332, 1433–1435. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Xu, S.; Mihaylova, M.M.; Zheng, B.; Hou, X.; Jiang, B.; Park, O.; Luo, Z.; Lefai, E.; Shyy, J.Y.; et al. AMPK phosphorylates and inhibits SREBP activity to attenuate hepatic steatosis and atherosclerosis in diet-induced insulin-resistant mice. Cell Metab. 2011, 13, 376–388. [Google Scholar] [CrossRef] [PubMed]
- Han, S.J.; Jang, H.S.; Noh, M.R.; Kim, J.; Kong, M.J.; Kim, J.I.; Park, J.W.; Park, K.M. Mitochondrial NADP(+)-Dependent Isocitrate Dehydrogenase Deficiency Exacerbates Mitochondrial and Cell Damage after Kidney Ischemia-Reperfusion Injury. J. Am. Soc. Nephrol. 2017, 28, 1200–1215. [Google Scholar] [CrossRef] [PubMed]
- Hardie, D.G.; Ross, F.A.; Hawley, S.A. AMP-activated protein kinase: A target for drugs both ancient and modern. Chem. Biol. 2012, 19, 1222–1236. [Google Scholar] [CrossRef]
- Senese, R.; Lasala, P.; Leanza, C.; de Lange, P. New avenues for regulation of lipid metabolism by thyroid hormones and analogs. Front. Physiol. 2014, 5, 475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Lange, P.; Cioffi, F.; Senese, R.; Moreno, M.; Lombardi, A.; Silvestri, E.; De Matteis, R.; Lionetti, L.; Mollica, M.P.; Goglia, F.; et al. Nonthyrotoxic prevention of diet-induced insulin resistance by 3,5-diiodo-L-thyronine in rats. Diabetes 2011, 60, 2730–2739. [Google Scholar] [CrossRef]
- Blennemann, B.; Leahy, P.; Kim, T.S.; Freake, H.C. Tissue-specific regulation of lipogenic mRNAs by thyroid hormone. Mol. Cell. Endocrinol. 1995, 110, 1–8. [Google Scholar] [CrossRef]
- Huang, C.; Freake, H.C. Thyroid hormone regulates the acetyl-CoA carboxylase PI promoter. Biochem. Biophys. Res. Commun. 1998, 249, 704–708. [Google Scholar] [CrossRef] [PubMed]
- Rubattu, S.; Pagliaro, B.; Pierelli, G.; Santolamazza, C.; Castro, S.D.; Mennuni, S.; Volpe, M. Pathogenesis of target organ damage in hypertension: Role of mitochondrial oxidative stress. Int. J. Mol. Sci. 2014, 16, 823–839. [Google Scholar] [CrossRef] [PubMed]
- Robertson, J.D.; Starnes, J.W.; Kehrer, J.P. Cosubstrates involved in the reduction of cytosolic glutathione disulfide in rat heart. Toxicology 1997, 124, 11–19. [Google Scholar] [CrossRef]
- Rabinovitch, R.C.; Samborska, B.; Faubert, B.; Ma, E.H.; Gravel, S.P.; Andrzejewski, S.; Raissi, T.C.; Pause, A.; St-Pierre, J.; Jones, R.G. AMPK Maintains Cellular Metabolic Homeostasis through Regulation of Mitochondrial Reactive Oxygen Species. Cell Rep. 2017, 21, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, X.X.; Leung, E.L.; Xie, Y.; Liu, Z.Q.; Zheng, Y.F.; Yao, X.J.; Lu, L.L.; Wu, J.L.; He, J.X.; Yuan, Z.W.; et al. Suppression of Lipogenesis via Reactive Oxygen Species-AMPK Signaling for Treating Malignant and Proliferative Diseases. Antioxid. Redox Signal. 2018, 28, 339–357. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Rank | Molecules in Network | Score 2 | Focused Molecule 3 | Top Functional Networks |
|---|---|---|---|---|
| 1 | AHCY, ALAS1, AMFR, Apoc3, C1S, C3, CAT, CEBPD, Ces2a, Clec2d, CLOCK, CXCL3, Cyp2c40, CYP2C9, CYP2E1, Cyp4a14, DHCR7, DNAJC7, EEF2K, FOS, G0S2, Gstm6, HES1, HMGCL, HSF2, ICAM1, IL1B, NFE2L1, NR1I2, PPARA, PPARGC1A, RXRA, ST3GAL5, SULT2A1, TXNIP | 21 | 17 | Lipid Metabolism, Molecular Transport, Small Molecule Biochemistry |
| 2 | AACS, ACACA, ACACB, AGPAT2, BTC, CCND3, CEBPA, Cyp2c12, DUSP1, ELOVL2, FABP5, FASN, FOS, HSD3B7, KLB, MBTPS1, MID1IP1, MLX, MLXIPL, NCOR1, NEUROG3, NFIL3, ONECUT1, PRKAA2, PSME3, RARG, RXRA, SREBF2, SRSF2, SULT2A1, THRSP, TKFC, TRIB1, Ugt1a7c, VDR | 19 | 16 | Lipid Metabolism, Molecular Transport, Small Molecule Biochemistry |
| 3 | ARNT, C3, Calm1, CEBPD, CIAPIN1, DDC, DNM2, EGF, EGR1, ESR1, FOS, GDF15, HAMP, HDAC3, ICAM1, IL6ST, JAK2, MAPK3, ME1, MTOR, NCOA1, PCNA, PDE4A, PPP2CB, PRKAR1A, RGS16, RGS3, RICTOR, RPS15, RPS4Y1, RPS6KB1, SDHB, SERPINA1, SP1, STAT3 | 19 | 16 | Cell Death and Survival, Cancer, Hematological Disease |
| 4 | DYNC1H1, S100A10 | 1 | 1 | Cellular Growth and Proliferation, Developmental Disorder, Hereditary Disorder |
| 5 | DLG1, GJB1 | 1 | 1 | Carbohydrate Metabolism, Cell-To-Cell Signaling and Interaction, Cellular Function and Maintenance |
| 6 | GATA1, MYCN | 1 | 1 | Cancer, Hematological Disease, Organismal Injury and Abnormalities |
| 7 | EIF2AK2, TUBA1A | 1 | 1 | Developmental Disorder, Hereditary Disorder, Neurological Disease |
| 8 | ADAM15, MBD2 | 1 | 1 | Cell-To-Cell Signaling and Interaction, Hair and Skin Development and Function, Cellular Compromise |
| 9 | PRKAA1, PRKAB1, PRKAG1 | 1 | 1 | Protein Synthesis, Cell Morphology, Cellular Function and Maintenance |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pan, J.H.; Tang, J.; Redding, M.C.; Beane, K.E.; Conner, C.L.; Cho, Y.J.; Zhao, J.; Kim, J.H.; Kong, B.C.; Lee, J.H.; et al. Hepatic Transcriptomics Reveals that Lipogenesis Is a Key Signaling Pathway in Isocitrate Dehydrogenase 2 Deficient Mice. Genes 2019, 10, 728. https://doi.org/10.3390/genes10090728
Pan JH, Tang J, Redding MC, Beane KE, Conner CL, Cho YJ, Zhao J, Kim JH, Kong BC, Lee JH, et al. Hepatic Transcriptomics Reveals that Lipogenesis Is a Key Signaling Pathway in Isocitrate Dehydrogenase 2 Deficient Mice. Genes. 2019; 10(9):728. https://doi.org/10.3390/genes10090728
Chicago/Turabian StylePan, Jeong Hoon, Jingsi Tang, Mersady C. Redding, Kaleigh E. Beane, Cara L. Conner, Yun Jeong Cho, Jiangchao Zhao, Jun Ho Kim, Byungwhi C. Kong, Jin Hyup Lee, and et al. 2019. "Hepatic Transcriptomics Reveals that Lipogenesis Is a Key Signaling Pathway in Isocitrate Dehydrogenase 2 Deficient Mice" Genes 10, no. 9: 728. https://doi.org/10.3390/genes10090728