Meta-Analysis of Cancer Triploidy: Rearrangements of Genome Complements in Male Human Tumors Are Characterized by XXY Karyotypes

 , and
, and
Abstract
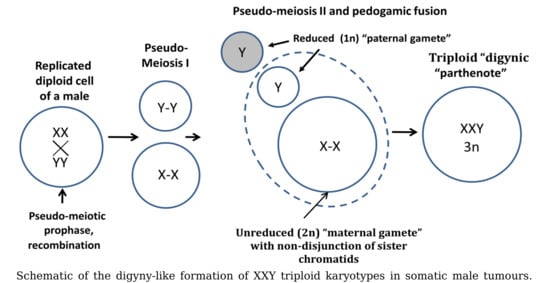
:
1. Introduction
2. Materials and Methods
3. Results
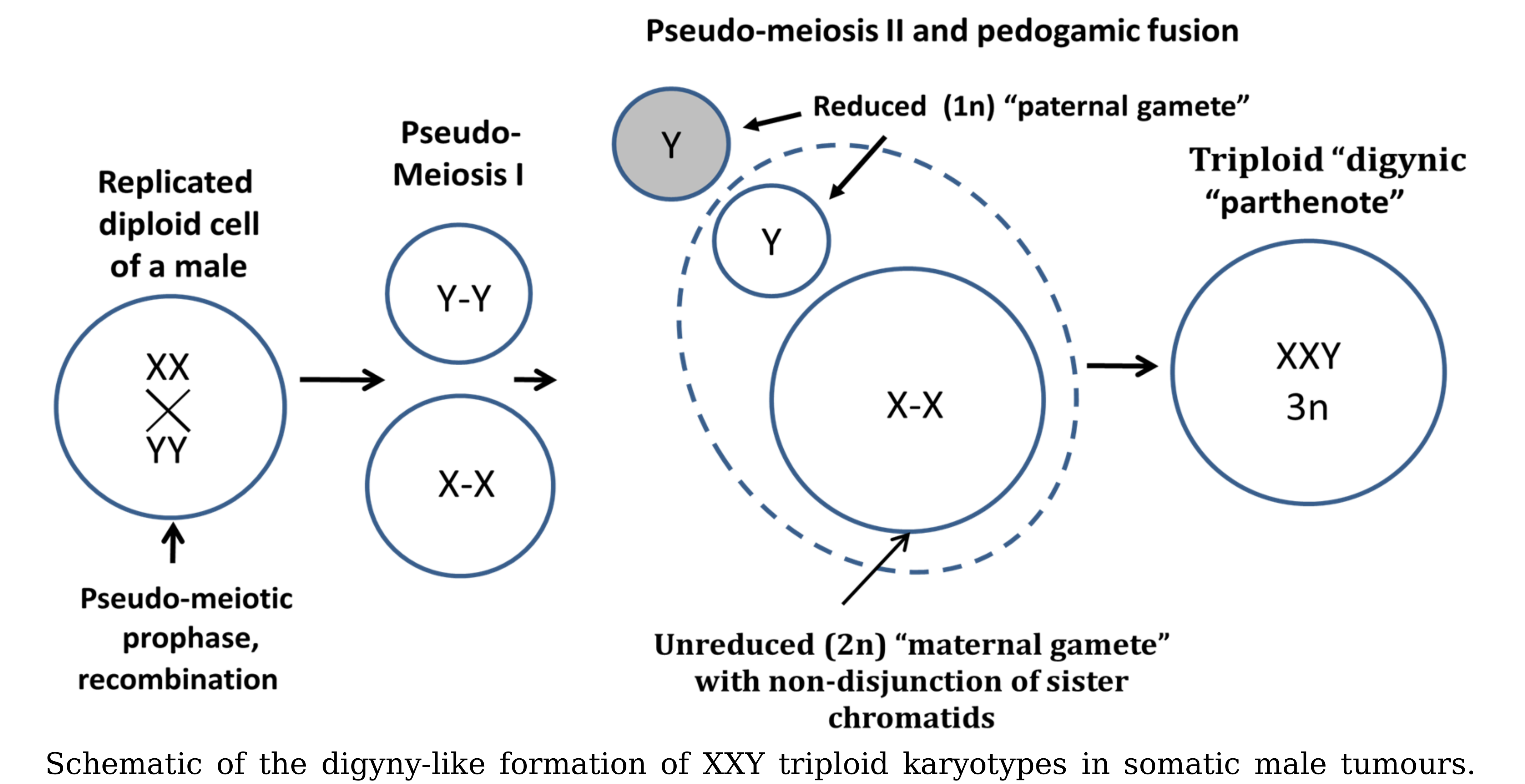
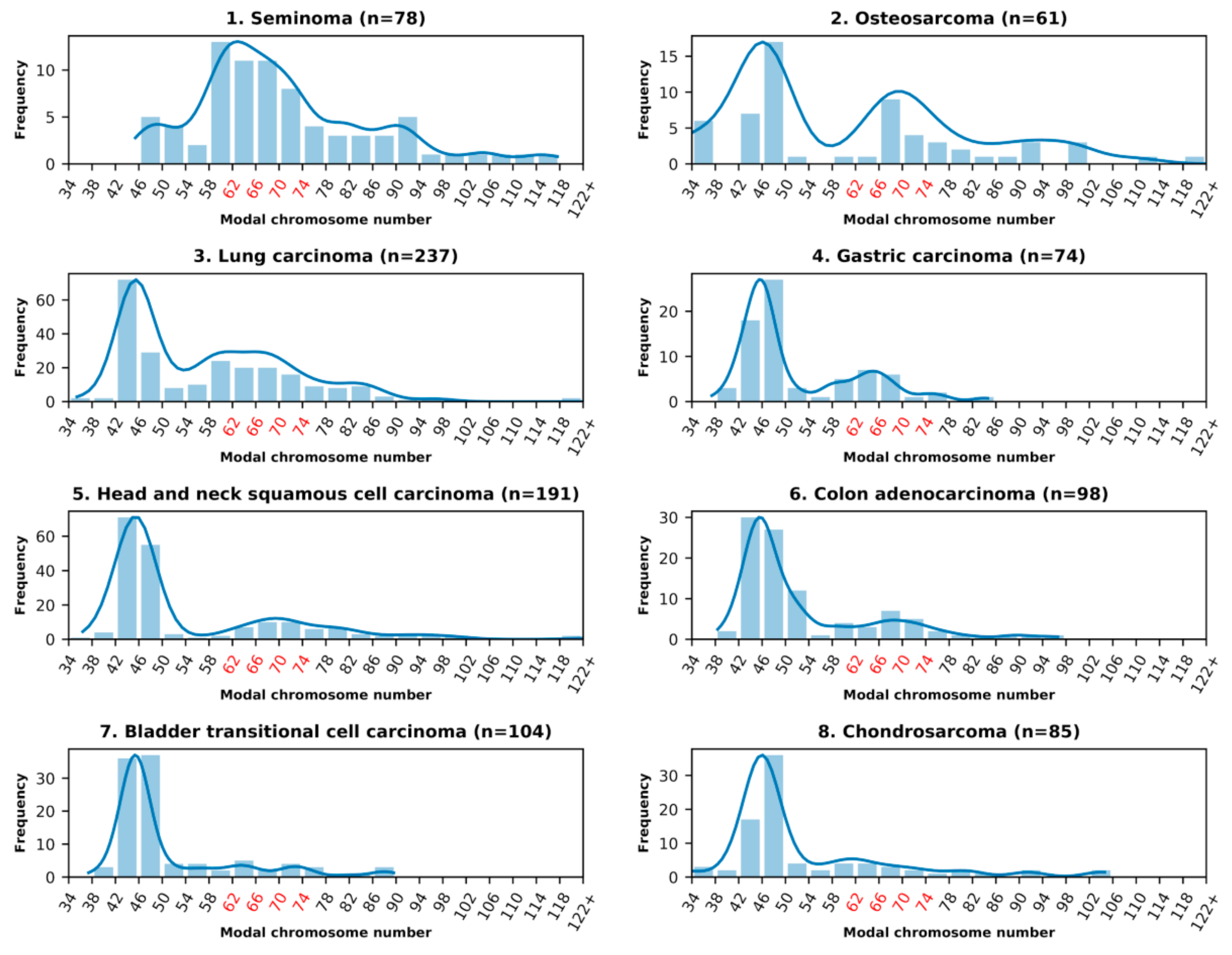
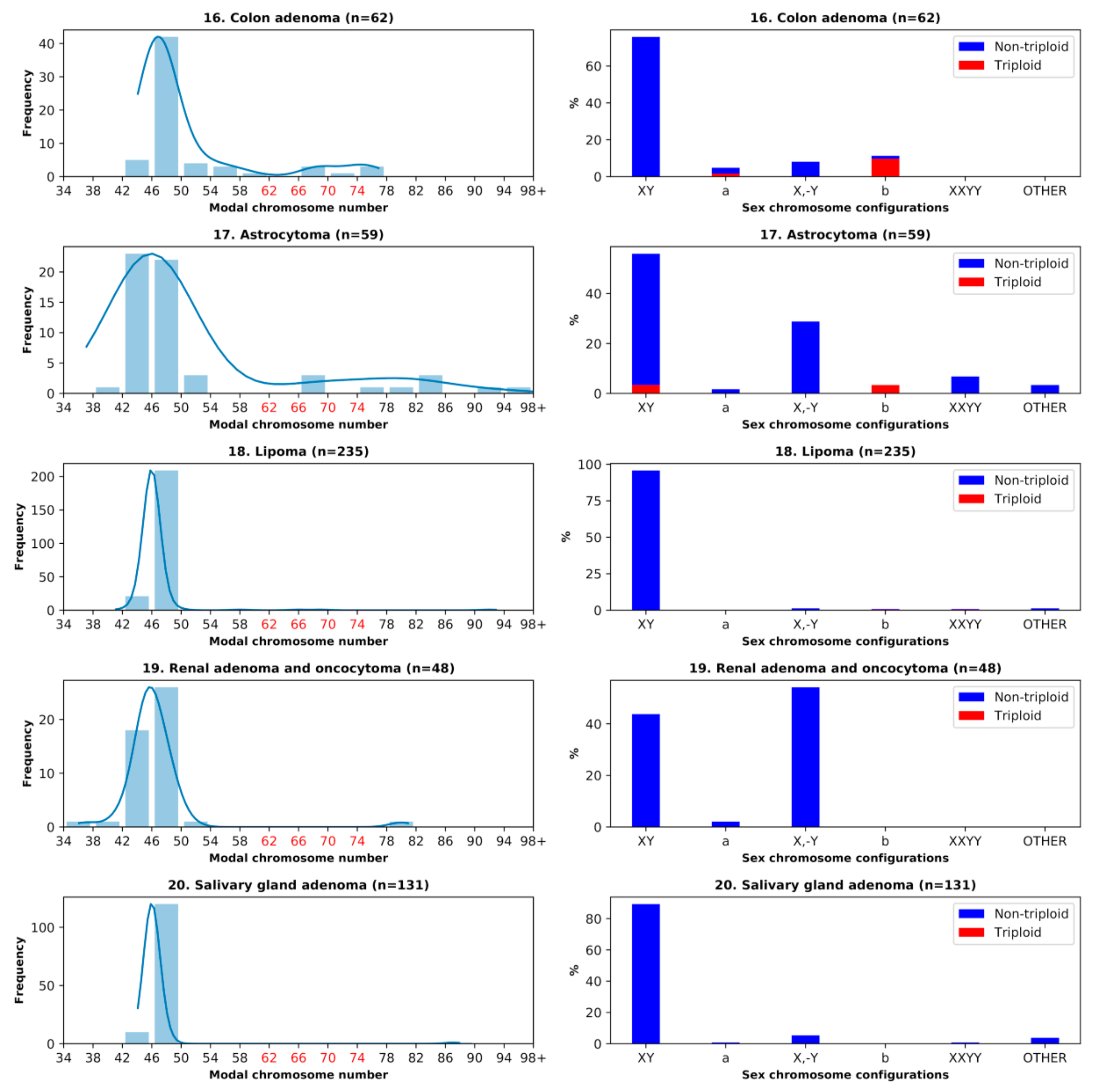
3.1. Analysis of the Histograms of the Modal Chromosome Numbers in 15 Cohorts of Malignant Tumors
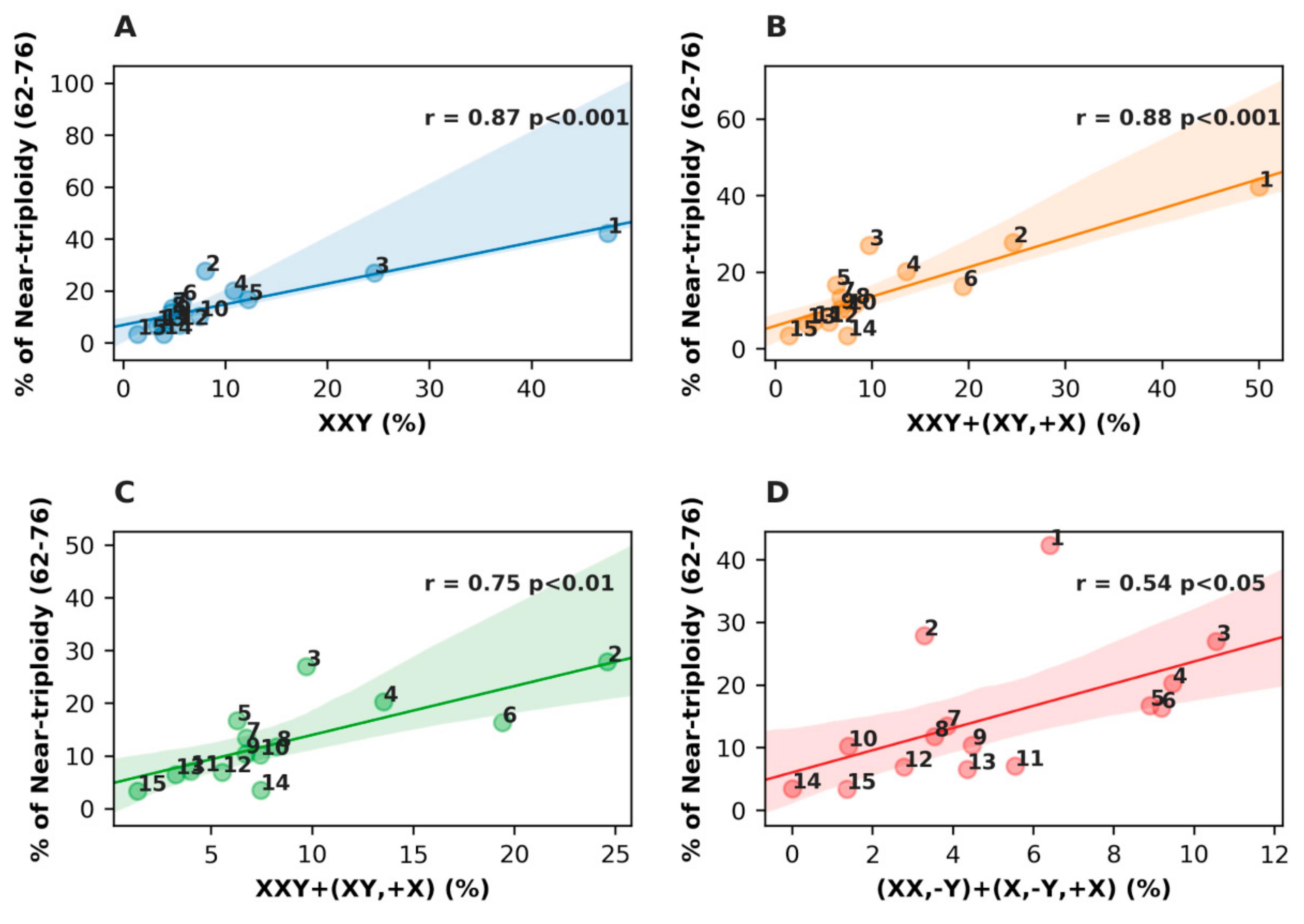
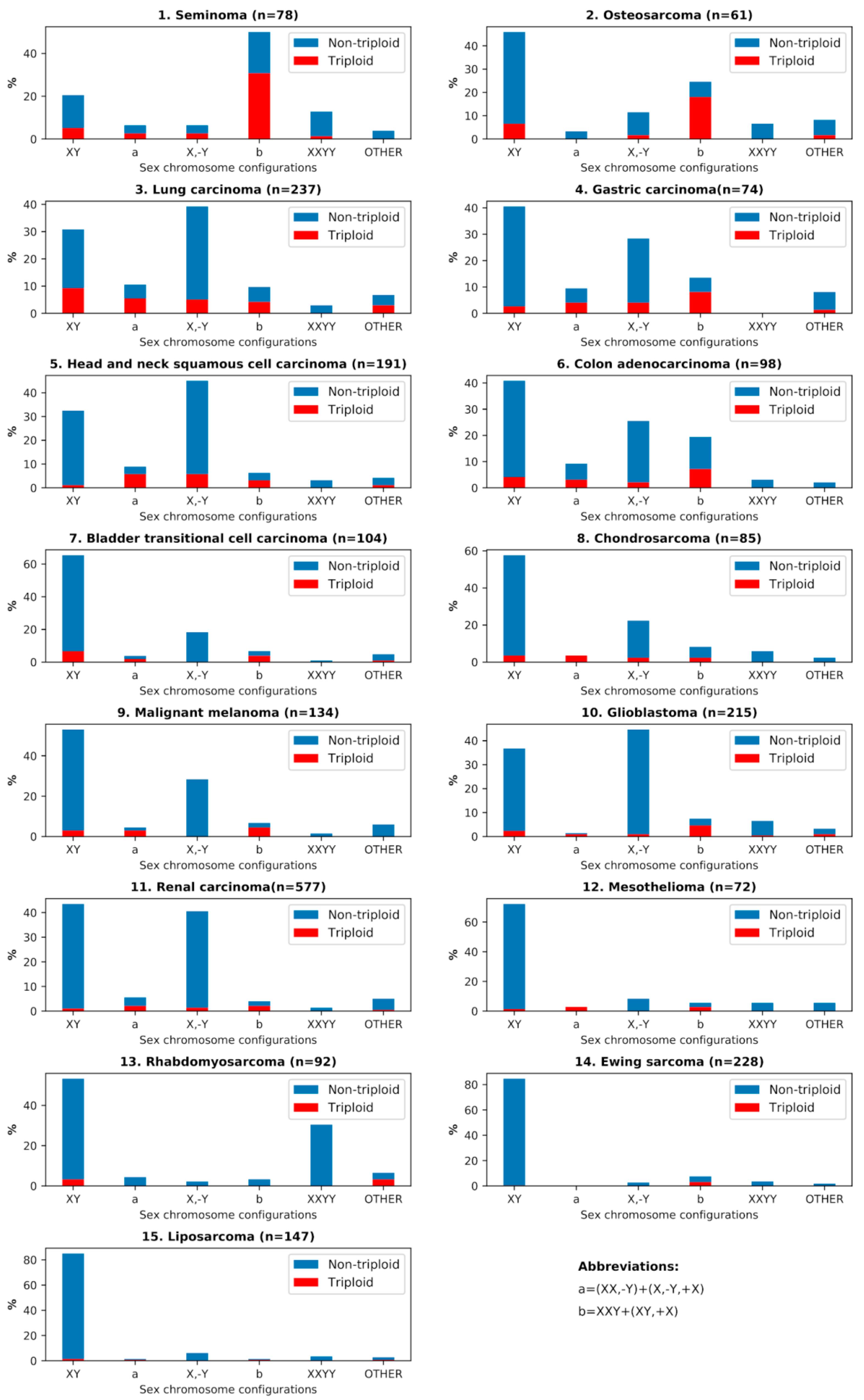
3.2. Analysis of the Sex Chromosome Sets with #X-Disomy in Each Malignant Tumor Cohort in Relation to Ploidy for their Karyotypes
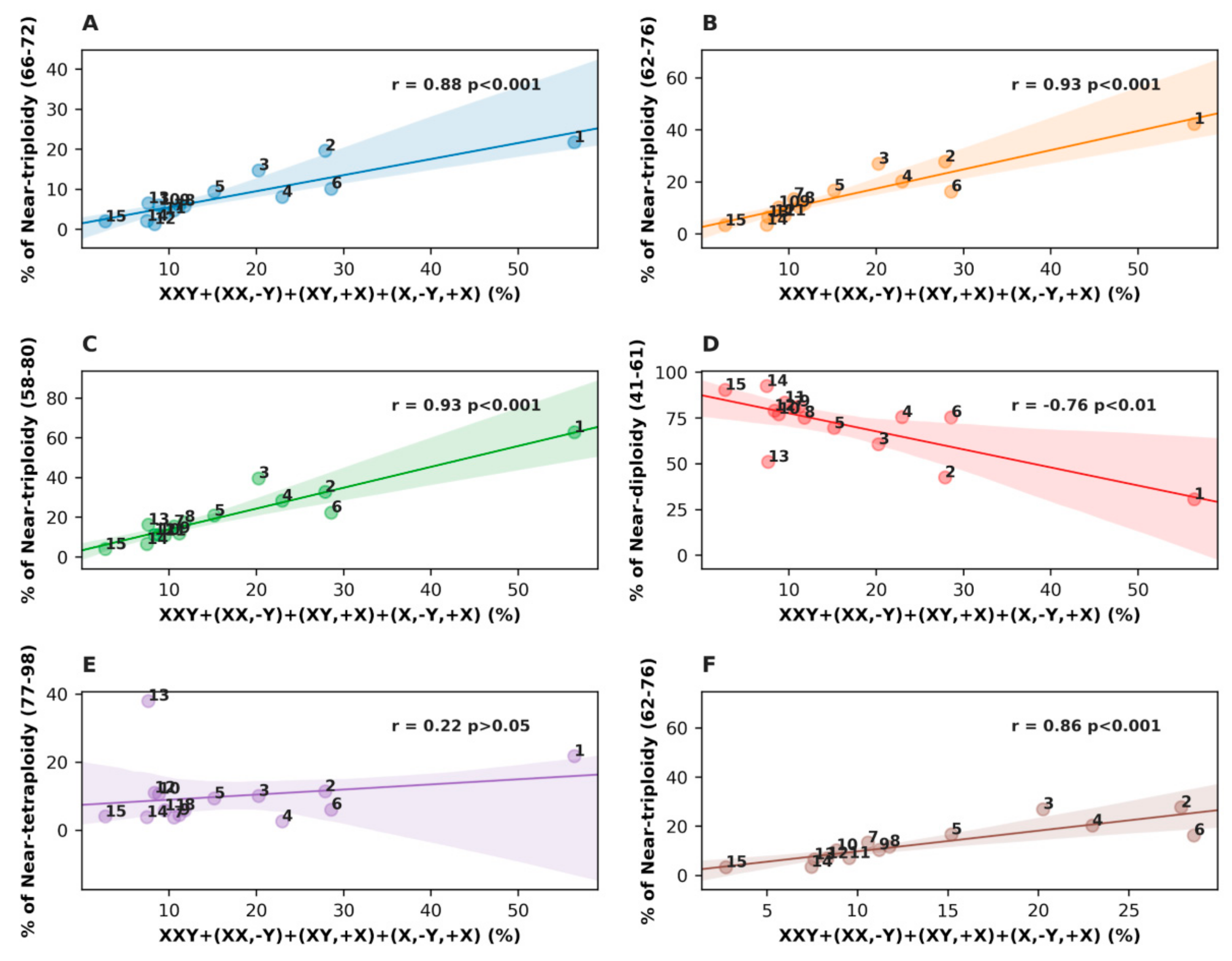
3.3. Analysis of All Sex Chromosome Configurations in Relation to Near-Triploidy in Malignant Tumors
3.4. Benign Tumors: Study of the Doubled X-Chromosome Karyotypes and Near-Triploidy
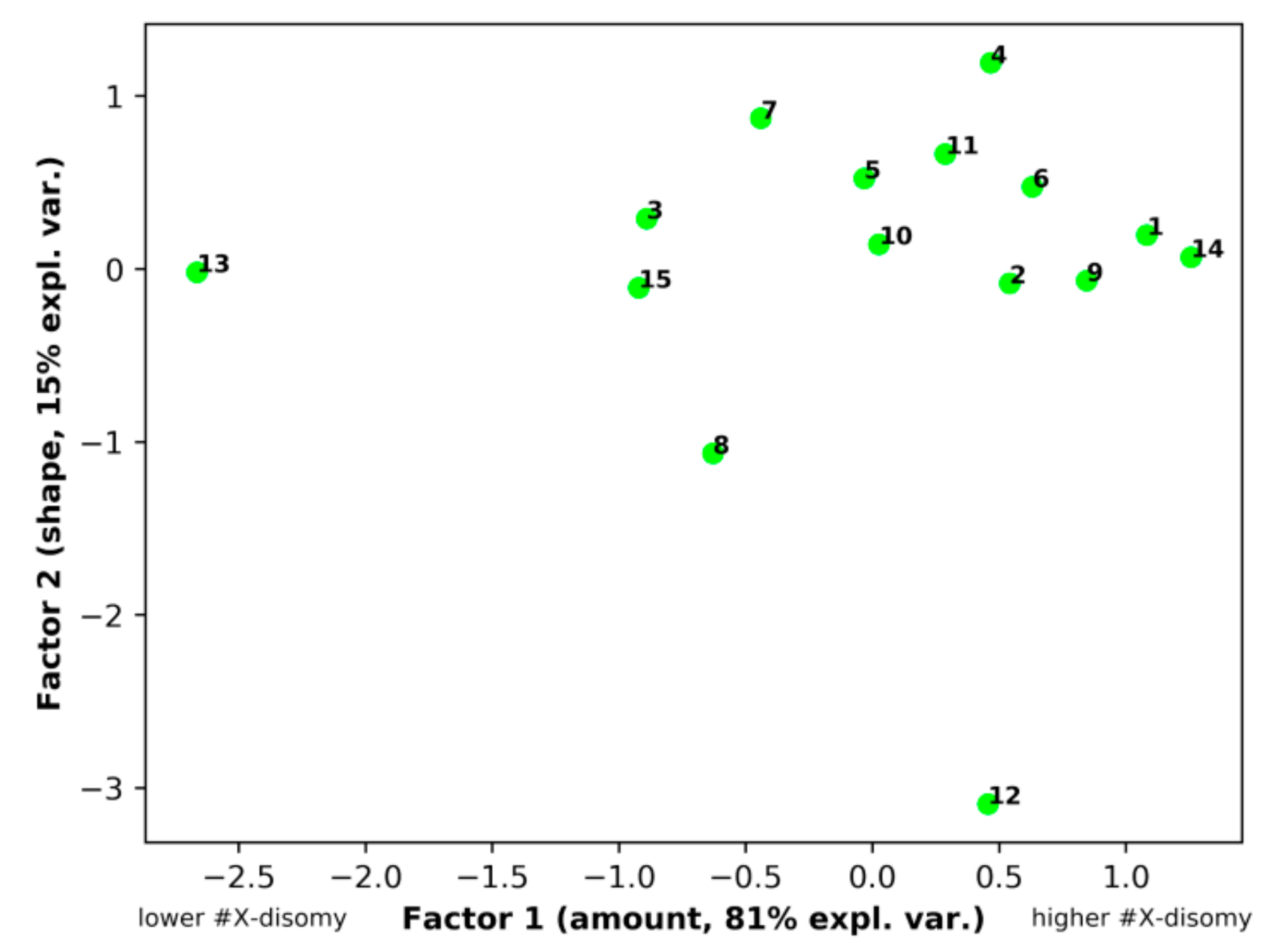
3.5. Principal Component Analysis (PCA)
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Holland, A.J.; Cleveland, D.W. Losing balance: The origin and impact of aneuploidy in cancer. EMBO Rep. 2012, 13, 501–514. [Google Scholar] [CrossRef] [PubMed]
- Pfau, S.J.; Amon, A. Chromosomal instability and aneuploidy in cancer: From yeast to man. EMBO Rep. 2012, 13, 515–527. [Google Scholar] [CrossRef] [PubMed]
- Pihan, G.A. Centrosome dysfunction contributes to chromosome instability, chromoanagenesis, and genome reprograming in cancer. Front. Oncol. 2013, 3, 277. [Google Scholar] [CrossRef] [PubMed]
- Heng, H.H.; Bremer, S.W.; Stevens, J.B.; Horne, S.D.; Liu, G.; Abdallah, B.Y.; Ye, K.J.; Ye, C.J. Chromosomal instability (CIN): What it is and why it is crucial to cancer evolution. Cancer Metastasis Rev. 2013, 32, 325–340. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Stevens, J.B.; Horne, S.D.; Abdallah, B.Y.; Ye, K.J.; Bremer, S.W.; Ye, C.J.; Chen, D.J.; Heng, H.H. Genome chaos: Survival strategy during crisis. Cell Cycle 2014, 13, 528–537. [Google Scholar] [CrossRef] [PubMed]
- Cross, W.C.; Graham, T.A.; Wright, N.A. New paradigms in clonal evolution: Punctuated equilibrium in cancer. J. Pathol. 2016, 240, 126–136. [Google Scholar] [CrossRef] [PubMed]
- Ly, P.; Cleveland, D.W. Rebuilding Chromosomes After Catastrophe: Emerging Mechanisms of Chromothripsis. Trends Cell Biol. 2017, 27, 917–930. [Google Scholar] [CrossRef]
- Ye, C.J.; Regan, S.; Liu, G.; Alemara, S.; Heng, H.H. Understanding aneuploidy in cancer through the lens of system inheritance, fuzzy inheritance and emergence of new genome systems. Mol. Cytogenet. 2018, 11, 31. [Google Scholar] [CrossRef]
- Weaver, B.A.A.; Silk, A.D.; Montagna, C.; Verdier-Pinard, P.; Cleveland, D.W. Aneuploidy acts both oncogenically and as a tumor suppressor. Cancer Cell 2007, 11, 25–36. [Google Scholar] [CrossRef]
- Sheltzer, J.M.; Ko, J.H.; Replogle, J.M.; Habibe Burgos, N.C.; Chung, E.S.; Meehl, C.M.; Sayles, N.M.; Passerini, V.; Storchova, Z.; Amon, A. Single-chromosome Gains Commonly Function as Tumor Suppressors. Cancer Cell 2017, 31, 240–255. [Google Scholar] [CrossRef] [Green Version]
- Andersson, D.I.; Hughes, D. Muller’s ratchet decreases fitness of a DNA-based microbe. Proc. Natl. Acad. Sci. USA 1996, 93, 906–907. [Google Scholar] [CrossRef] [PubMed]
- Demin, S.Y.; Berdieva, M.A.; Goodkov, A.V. Cyclic Polyploidy in Obligate Agamic Amoebae. Cell Tissue Biol. 2019, 13, 242–246. [Google Scholar] [CrossRef]
- Raikov, I.B. The Protozoan Nucleus; Cell Biology Monographs; Springer: Wien, Austria, 1982; Volume 9, ISBN 9783709141380. [Google Scholar]
- Maciver, S.K. Asexual Amoebae Escape Muller’s Ratchet through Polyploidy. Trends Parasitol. 2016, 32, 855–862. [Google Scholar] [CrossRef] [PubMed]
- Vincent, M.D. Cancer: Beyond speciation. Adv. Cancer Res. 2011, 112, 283–350. [Google Scholar]
- Trigos, A.S.; Pearson, R.B.; Papenfuss, A.T.; Goode, D.L. Altered interactions between unicellular and multicellular genes drive hallmarks of transformation in a diverse range of solid tumors. Proc. Natl. Acad. Sci. USA 2017, 114, 6406–6411. [Google Scholar] [CrossRef] [Green Version]
- Vinogradov, A.E.; Anatskaya, O.V. Evolutionary framework of the human interactome: Unicellular and multicellular giant clusters. Biosystems 2019, 181, 82–87. [Google Scholar] [CrossRef]
- Vazquez-Martin, A.; Anatskaya, O.V.; Giuliani, A.; Erenpreisa, J.; Huang, S.; Salmina, K.; Inashkina, I.; Huna, A.; Nikolsky, N.N.; Vinogradov, A.E. Somatic polyploidy is associated with the upregulation of c-MYC interacting genes and EMT-like signature. Oncotarget 2016, 7, 75235–75260. [Google Scholar] [CrossRef] [Green Version]
- Erenpreisa, J.; Giuliani, A.; Vinogradov, A.E.; Anatskaya, O.V.; Vazquez-Martin, A.; Salmina, K.; Cragg, M.S. Stress-induced polyploidy shifts somatic cells towards a pro-tumourogenic unicellular gene transcription network. Cancer Hypotheses 2018, 1, 1–20. [Google Scholar]
- Anatskaya, O.V.; Erenpreisa, J.; Giuliani, A.; Salmina, K.; Vinogradov, A.E. Polyploidy reprograms regulatory pathways towards unicellular mode: The role in stress response, drug resistance, growth and cancer. Proc. Biopolym. Cell 2019, 35, 2019. [Google Scholar]
- Notta, F.; Chan-Seng-Yue, M.; Lemire, M.; Li, Y.; Wilson, G.W.; Connor, A.A.; Denroche, R.E.; Liang, S.-B.; Brown, A.M.K.; Kim, J.C.; et al. A renewed model of pancreatic cancer evolution based on genomic rearrangement patterns. Nature 2016, 538, 378–382. [Google Scholar] [CrossRef]
- Salmina, K.; Jankevics, E.; Huna, A.; Perminov, D.; Radovica, I.; Klymenko, T.; Ivanov, A.; Jascenko, E.; Scherthan, H.; Cragg, M.; et al. Up-regulation of the embryonic self-renewal network through reversible polyploidy in irradiated p53-mutant tumour cells. Exp. Cell Res. 2010, 316, 2099–2112. [Google Scholar] [CrossRef]
- Kondrashov, A.S. Evolutionary Genetics of Life Cycles. Annu. Rev. Ecol. Syst. 1997, 28, 391–435. [Google Scholar] [CrossRef]
- Kondrashov, A.S. The asexual ploidy cycle and the origin of sex. Nature 1994, 370, 213–216. [Google Scholar] [CrossRef]
- Rajaraman, R.; Rajaraman, M.M.; Rajaraman, S.R.; Guernsey, D.L. Neosis—A paradigm of self-renewal in cancer. Cell Biol. Int. 2005, 29, 1084–1097. [Google Scholar] [CrossRef]
- Erenpreisa, J.; Cragg, M.S. Cancer: A matter of life cycle? Cell Biol. Int. 2007, 31, 1507–1510. [Google Scholar] [CrossRef]
- Erenpreisa, J.; Cragg, M.S. MOS, aneuploidy and the ploidy cycle of cancer cells. Oncogene 2010, 29, 5447–5451. [Google Scholar] [CrossRef] [Green Version]
- Erenpreisa, J.; Kalejs, M.; Cragg, M.S. Mitotic catastrophe and endomitosis in tumour cells: An evolutionary key to a molecular solution. Cell Biol. Int. 2005, 29, 1012–1018. [Google Scholar] [CrossRef]
- Gerashchenko, B.I.; Salmina, K.; Eglitis, J.; Huna, A.; Grjunberga, V.; Erenpreisa, J. Disentangling the aneuploidy and senescence paradoxes: A study of triploid breast cancers non-responsive to neoadjuvant therapy. Histochem. Cell Biol. 2016, 145, 497–508. [Google Scholar] [CrossRef]
- Schulze, S.; Petersen, I. Gender and ploidy in cancer survival. Cell. Oncol. 2011, 34, 199–208. [Google Scholar] [CrossRef] [Green Version]
- Di Oto, E.; Monti, V.; Cucchi, M.C.; Masetti, R.; Varga, Z.; Foschini, M.P. X chromosome gain in male breast cancer. Hum. Pathol. 2015, 46, 1908–1912. [Google Scholar] [CrossRef]
- Yamamoto, K.; Nagata, K.; Kida, A.; Hamaguchi, H. Acquired gain of an X chromosome as the sole abnormality in the blast crisis of chronic neutrophilic leukemia. Cancer Genet. Cytogenet. 2002, 134, 84–87. [Google Scholar] [CrossRef]
- Okada, Y.; Nishikawa, R.; Matsutani, M.; Louis, D.N. Hypomethylated X chromosome gain and rare isochromosome 12p in diverse intracranial germ cell tumors. J. Neuropathol. Exp. Neurol. 2002, 61, 531–538. [Google Scholar] [CrossRef]
- Hunter, S.; Gramlich, T.; Abbott, K.; Varma, V. Y chromosome loss in esophageal carcinoma: An in situ hybridization study. Genes Chromosomes Cancer 1993, 8, 172–177. [Google Scholar] [CrossRef]
- Park, S.-J.; Jeong, S.-Y.; Kim, H.J. Y chromosome loss and other genomic alterations in hepatocellular carcinoma cell lines analyzed by CGH and CGH array. Cancer Genet. Cytogenet. 2006, 166, 56–64. [Google Scholar] [CrossRef]
- Bianchi, N.O. Y chromosome structural and functional changes in human malignant diseases. Mutat. Res. 2009, 682, 21–27. [Google Scholar] [CrossRef]
- Duijf, P.H.G.; Schultz, N.; Benezra, R. Cancer cells preferentially lose small chromosomes. Int. J. Cancer 2013, 132, 2316–2326. [Google Scholar] [CrossRef]
- Castedo, S.M.; de Jong, B.; Oosterhuis, J.W.; Seruca, R.; te Meerman, G.J.; Dam, A.; Schraffordt Koops, H. Cytogenetic analysis of ten human seminomas. Cancer Res. 1989, 49, 439–443. [Google Scholar]
- Mitelman, F.; Johansson, B.; Mertens, F. Mitelman Database of Chromosome Aberrations and Gene Fusions in Cancer. Available online: http://cgap.nci.nih.gov/Chromosomes/Mitelman (accessed on 18 January 2019).
- Onaitis, M.; Hanna, J. Cell of origin of lung cancer. J. Carcinog. 2013, 12, 6. [Google Scholar] [CrossRef]
- Oliphant, T.E. A guide to NumPy; Trelgol Publishing: Spanish Fork, UT, USA, 2006. [Google Scholar]
- McKinney, W. Data Structures for Statistical Computing in Python. In Proceedings of the 9th Python in Science Conference, Austin, TX, USA, 28 June–3 July 2010; van der Walt, S., Millman, J., Eds.; pp. 51–56. [Google Scholar]
- Jones, E.; Oliphant, T.; Peterson, P. SciPy: Open Source Scientific Tools for Python. Available online: http://www.scipy.org/ (accessed on 18 January 2019).
- Shaffer, L.G.; McGowan-Jordan, J.; Schmid, M. International Standing Committee on Human Cytogenetic Nomenclature. In ISCN 2013: An International System for Human Cytogenetic Nomenclature; Shaffer, L.G., McGowan-Jordan, J., Schmid, M., Eds.; Karger Medical and Scientific Publishers: Basel, Switzerland, 2013; ISBN 9783318022537. [Google Scholar]
- Jolicoeur, P.; Mosimann, J.E. Size and shape variation in the painted turtle. A principal component analysis. Growth 1960, 24, 339–354. [Google Scholar]
- Bickel, P.J.; Doksum, K.A. Mathematical Statistics: Basic Ideas and Selected Topics; Holden-Day: San Francisco, CA, USA, 1977. [Google Scholar]
- Giuliani, A. The application of principal component analysis to drug discovery and biomedical data. Drug Discov. Today 2017, 22, 1069–1076. [Google Scholar] [CrossRef]
- Herrick, S.E.; Mutsaers, S.E. Mesothelial progenitor cells and their potential in tissue engineering. Int. J. Biochem. Cell Biol. 2004, 36, 621–642. [Google Scholar] [CrossRef]
- Ozery-Flato, M.; Linhart, C.; Trakhtenbrot, L.; Izraeli, S.; Shamir, R. Large-scale analysis of chromosomal aberrations in cancer karyotypes reveals two distinct paths to aneuploidy. Genome Biol. 2011, 12, R61. [Google Scholar] [CrossRef]
- Nasmyth, K. Disseminating the genome: Joining, resolving, and separating sister chromatids during mitosis and meiosis. Annu. Rev. Genet. 2001, 35, 673–745. [Google Scholar] [CrossRef]
- Erenpreisa, J.; Cragg, M.S. Three steps to the immortality of cancer cells: Senescence, polyploidy and self-renewal. Cancer Cell Int. 2013, 13, 92. [Google Scholar] [CrossRef]
- Erenpreiss, J. Current Concepts of Malignant Growth. Part A: From A Normal Cell to Cancer; Zvaigzne: Riga, Latvia, 1993. [Google Scholar]
- Kalejs, M.; Ivanov, A.; Plakhins, G.; Cragg, M.S.; Emzinsh, D.; Illidge, T.M.; Erenpreisa, J. Upregulation of meiosis-specific genes in lymphoma cell lines following genotoxic insult and induction of mitotic catastrophe. BMC Cancer 2006, 6, 6. [Google Scholar] [CrossRef] [Green Version]
- Ianzini, F.; Kosmacek, E.A.; Nelson, E.S.; Napoli, E.; Erenpreisa, J.; Kalejs, M.; Mackey, M.A. Activation of meiosis-specific genes is associated with depolyploidization of human tumor cells following radiation-induced mitotic catastrophe. Cancer Res. 2009, 69, 2296–2304. [Google Scholar] [CrossRef]
- Vitale, I.; Senovilla, L.; Jemaà, M.; Michaud, M.; Galluzzi, L.; Kepp, O.; Nanty, L.; Criollo, A.; Rello-Varona, S.; Manic, G.; et al. Multipolar mitosis of tetraploid cells: Inhibition by p53 and dependency on Mos. EMBO J. 2010, 29, 1272–1284. [Google Scholar] [CrossRef]
- Gorgoulis, V.G.; Zacharatos, P.; Mariatos, G.; Liloglou, T.; Kokotas, S.; Kastrinakis, N.; Kotsinas, A.; Athanasiou, A.; Foukas, P.; Zoumpourlis, V.; et al. Deregulated expression of c-mos in non-small cell lung carcinomas: Relationship with p53 status, genomic instability, and tumor kinetics. Cancer Res. 2001, 61, 538–549. [Google Scholar]
- Erenpreisa, J.; Cragg, M.S.; Salmina, K.; Hausmann, M.; Scherthan, H. The role of meiotic cohesin REC8 in chromosome segregation in γ irradiation-induced endopolyploid tumour cells. Exp. Cell Res. 2009, 315, 2593–2603. [Google Scholar] [CrossRef]
- Chung, J.-Y.; Kitano, H.; Takikita, M.; Cho, H.; Noh, K.H.; Kim, T.W.; Ylaya, K.; Hanaoka, J.; Fukuoka, J.; Hewitt, S.M. Synaptonemal complex protein 3 as a novel prognostic marker in early stage non-small cell lung cancer. Hum. Pathol. 2013, 44, 472–479. [Google Scholar] [CrossRef]
- Lindsey, S.F.; Byrnes, D.M.; Eller, M.S.; Rosa, A.M.; Dabas, N.; Escandon, J.; Grichnik, J.M. Potential role of meiosis proteins in melanoma chromosomal instability. J. Skin Cancer 2013, 2013, 190109. [Google Scholar] [CrossRef]
- Wang, L.; Cao, J.; Ji, P.; Zhang, D.; Ma, L.; Dym, M.; Yu, Z.; Feng, L. Oocyte-like cells induced from mouse spermatogonial stem cells. Cell Biosci. 2012, 2, 27. [Google Scholar] [CrossRef]
- Erenpreisa, J.; Cragg, M.S. Mitotic death: A mechanism of survival? A review. Cancer Cell Int. 2001, 1, 1. [Google Scholar] [CrossRef]
- Zhang, C.-Z.; Spektor, A.; Cornils, H.; Francis, J.M.; Jackson, E.K.; Liu, S.; Meyerson, M.; Pellman, D. Chromothripsis from DNA damage in micronuclei. Nature 2015, 522, 179–184. [Google Scholar] [CrossRef] [Green Version]
- Mc Gee, M.M. Targeting the Mitotic Catastrophe Signaling Pathway in Cancer. Mediat. Inflamm. 2015, 2015, 146282. [Google Scholar] [CrossRef]
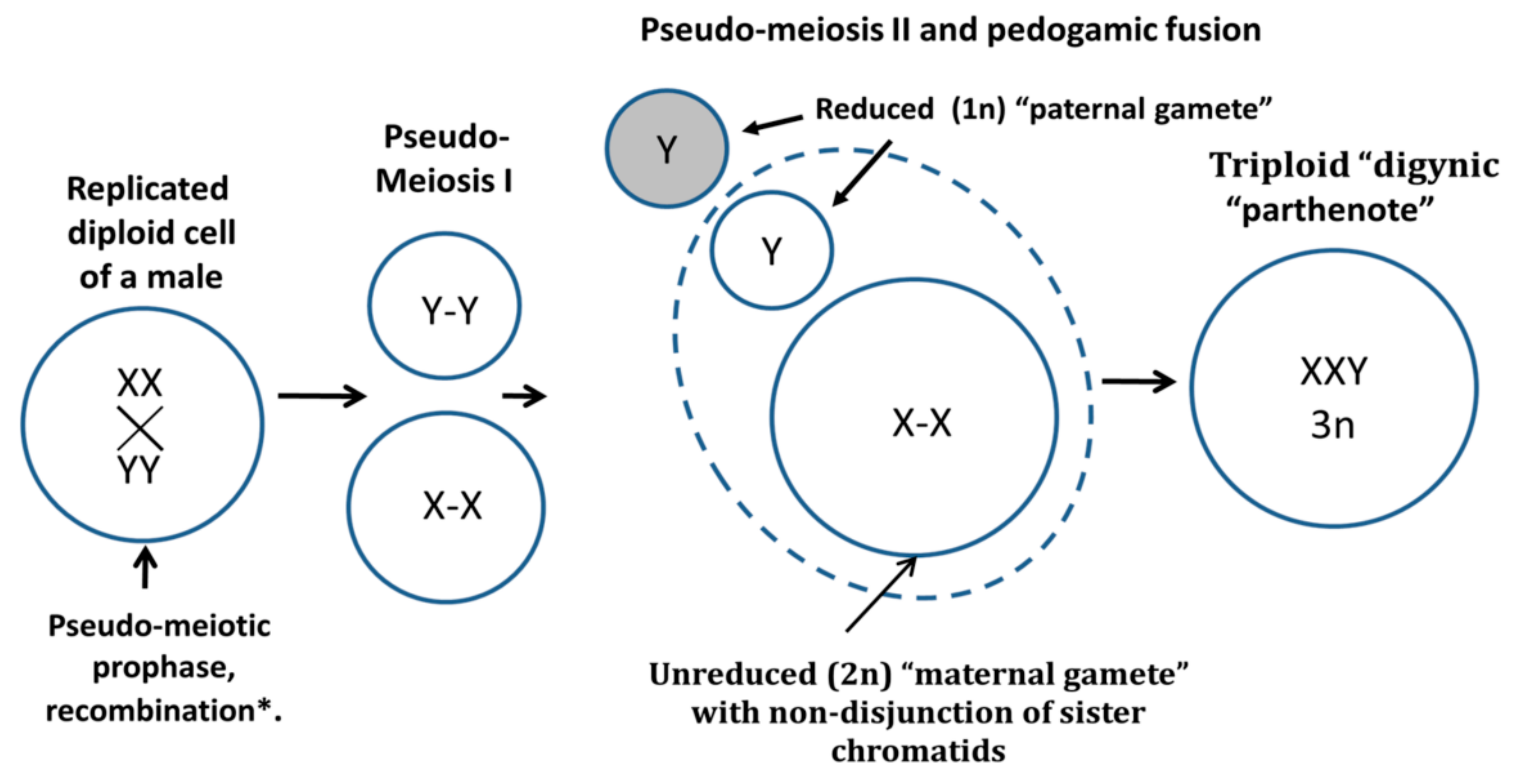
- Salmina, K.; Gerashchenko, B.I.; Hausmann, M.; Vainshelbaum, N.M.; Zayakin, P.; Erenpreiss, J.; Freivalds, T.; Cragg, M.S.; Erenpreisa, J. When Three Isn’t a Crowd: A Digyny Concept for Treatment-Resistant, Near-Triploid Human Cancers. Genes 2019, 10, 551. [Google Scholar] [CrossRef]
- Cutcutache, I.; Suzuki, Y.; Tan, I.B.; Ramgopal, S.; Zhang, S.; Ramnarayanan, K.; Gan, A.; Lee, H.H.; Tay, S.T.; Ooi, A.; et al. Exome-wide Sequencing Shows Low Mutation Rates and Identifies Novel Mutated Genes in Seminomas. Eur. Urol. 2015, 68, 77–83. [Google Scholar] [CrossRef] [Green Version]
- Armaghany, T.; Wilson, J.D.; Chu, Q.; Mills, G. Genetic alterations in colorectal cancer. Gastrointest. Cancer Res. 2012, 5, 19–27. [Google Scholar]
- Giaretti, W.; Venesio, T.; Sciutto, A.; Prevosto, C.; Geido, E.; Risio, M. Near-diploid and near-triploid human sporadic colorectal adenocarcinomas differ for KRAS2 and TP53 mutational status. Genes Chromosomes Cancer 2003, 37, 207–213. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Nº | Malignant Tumor Type | Number of Karyotypes | % of Near- Triploidy (62–76) | XXY % | XX,-Y % | (XY,+X)+(X,-Y,+X) % | XXY,+Y % |
|---|---|---|---|---|---|---|---|
| 1 | Seminoma | 78 | 42.31 | 47.44 | 3.85 | 5.13 | 10.26 |
| 2 | Osteosarcoma | 61 | 27.87 | 24.59 | 3.28 | 0.00 | 6.56 |
| 3 | Lung carcinoma | 237 | 27.00 | 8.02 | 9.70 | 2.53 | 2.53 |
| 4 | Gastric carcinoma | 74 | 20.27 | 10.81 | 5.41 | 6.76 | 1.35 |
| 5 | Head and neck squamous cell carcinoma | 191 | 16.75 | 5.76 | 8.90 | 0.52 | 1.57 |
| 6 | Colon adenocarcinoma | 98 | 16.33 | 12.24 | 6.12 | 10.20 | 6.12 |
| 7 | Transitional cell carcinoma | 104 | 13.46 | 4.81 | 3.85 | 1.92 | 1.92 |
| 8 | Chondrosarcoma | 85 | 11.76 | 4.71 | 3.53 | 3.53 | 0.00 |
| 9 | Malignant melanoma | 134 | 10.45 | 5.22 | 3.73 | 2.24 | 2.24 |
| 10 | Glioblastoma | 215 | 10.23 | 7.44 | 1.40 | 0.00 | 1.40 |
| 11 | Renal carcinoma | 577 | 7.11 | 3.81 | 4.68 | 1.04 | 1.21 |
| 12 | Mesothelioma | 72 | 6.94 | 5.56 | 2.78 | 0.00 | 2.78 |
| 13 | Rhabdomyosarcoma | 92 | 6.52 | 3.26 | 1.09 | 3.26 | 0.00 |
| 14 | Ewing sarcoma | 228 | 3.51 | 3.95 | 0.00 | 3.51 | 0.88 |
| 15 | Liposarcoma | 147 | 3.40 | 1.36 | 1.36 | 0.00 | 0.00 |
| Benign tumor type | |||||||
| 16 | Colon adenoma | 62 | 11.29 | 11.29 | 4.84 | 0.00 | 0.00 |
| 17 | Astrocytoma | 59 | 6.78 | 1.69 | 1.69 | 1.69 | 1.69 |
| 18 | Lipoma | 235 | 0.85 | 0.85 | 0.00 | 0.00 | 0.43 |
| 19 | Renal adenoma and oncocytoma | 48 | 0.00 | 0.00 | 2.08 | 0.00 | 0.00 |
| 20 | Salivary gland adenoma | 131 | 0.00 | 0.00 | 0.76 | 0.00 | 0.00 |
| Original Variables | Factor1 | Factor2 |
|---|---|---|
| #X disomy_62-76 | 0.96650 | −0.23119 |
| #X disomy_58-80 | 0.94894 | −0.05406 |
| XXY+XX,-Y_62-76 | 0.95938 | −0.23271 |
| #X disomy_66-72 | 0.69858 | 0.71287 |
| % of Explained Variance | 81.1 | 15.5 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vainshelbaum, N.M.; Zayakin, P.; Kleina, R.; Giuliani, A.; Erenpreisa, J. Meta-Analysis of Cancer Triploidy: Rearrangements of Genome Complements in Male Human Tumors Are Characterized by XXY Karyotypes. Genes 2019, 10, 613. https://doi.org/10.3390/genes10080613
Vainshelbaum NM, Zayakin P, Kleina R, Giuliani A, Erenpreisa J. Meta-Analysis of Cancer Triploidy: Rearrangements of Genome Complements in Male Human Tumors Are Characterized by XXY Karyotypes. Genes. 2019; 10(8):613. https://doi.org/10.3390/genes10080613
Chicago/Turabian StyleVainshelbaum, Ninel M., Pawel Zayakin, Regina Kleina, Alessandro Giuliani, and Jekaterina Erenpreisa. 2019. "Meta-Analysis of Cancer Triploidy: Rearrangements of Genome Complements in Male Human Tumors Are Characterized by XXY Karyotypes" Genes 10, no. 8: 613. https://doi.org/10.3390/genes10080613