DNILMF-LDA: Prediction of lncRNA-Disease Associations by Dual-Network Integrated Logistic Matrix Factorization and Bayesian Optimization

Abstract
:1. Introduction
2. Materials
2.1. lncRNA–Disease Associations Matrix
2.2. lncRNA Expression Similarity Matrix and Disease Semantic Similarity Matrix
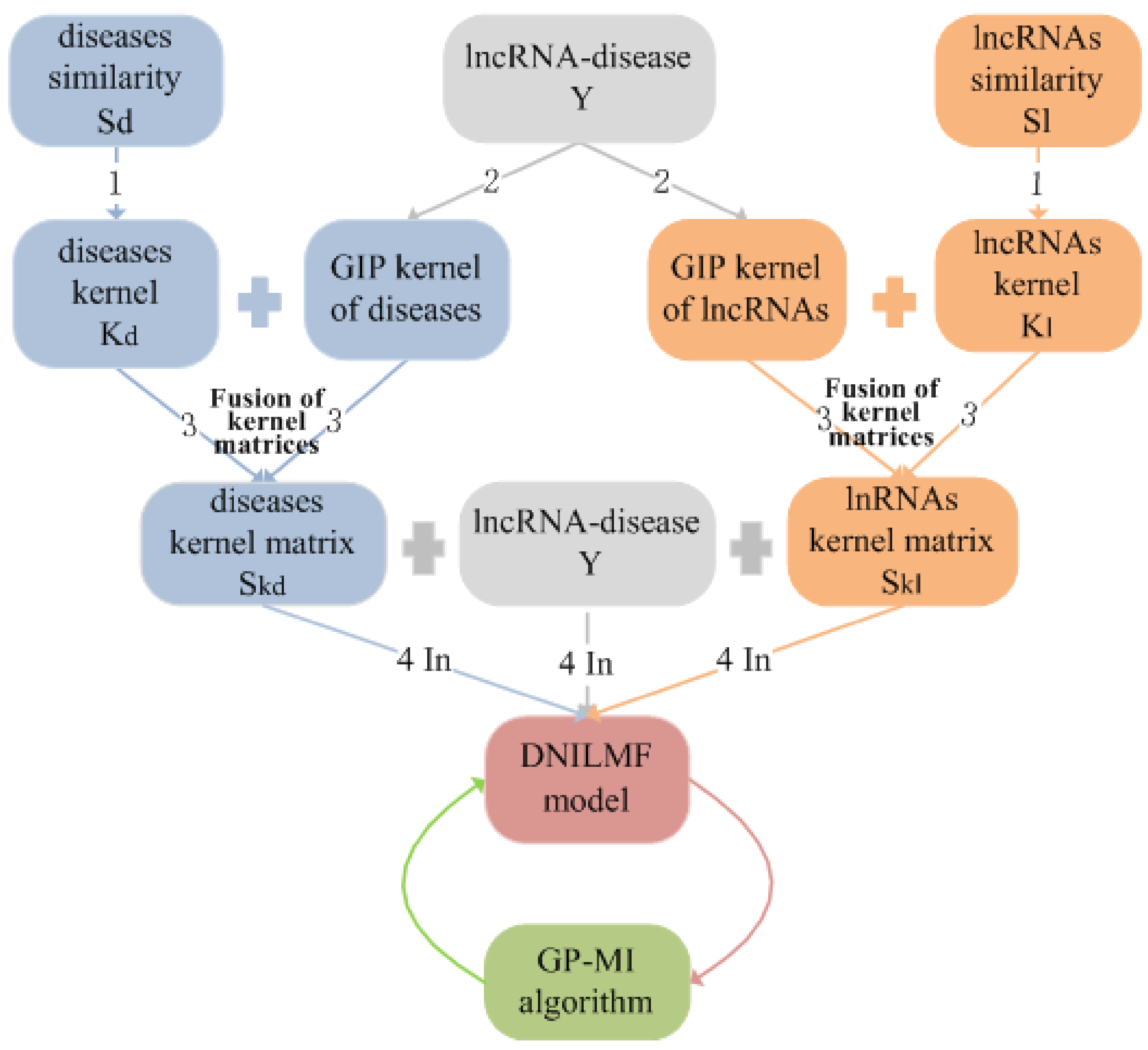
2.3. Similarity Kernel Matrices
2.4. Fusion of Similarity Kernel Matrices
3. Methods
3.1. Problem Formalization
3.2. Prediction of lncRNA–Disease Associations Using the DNILMF Model
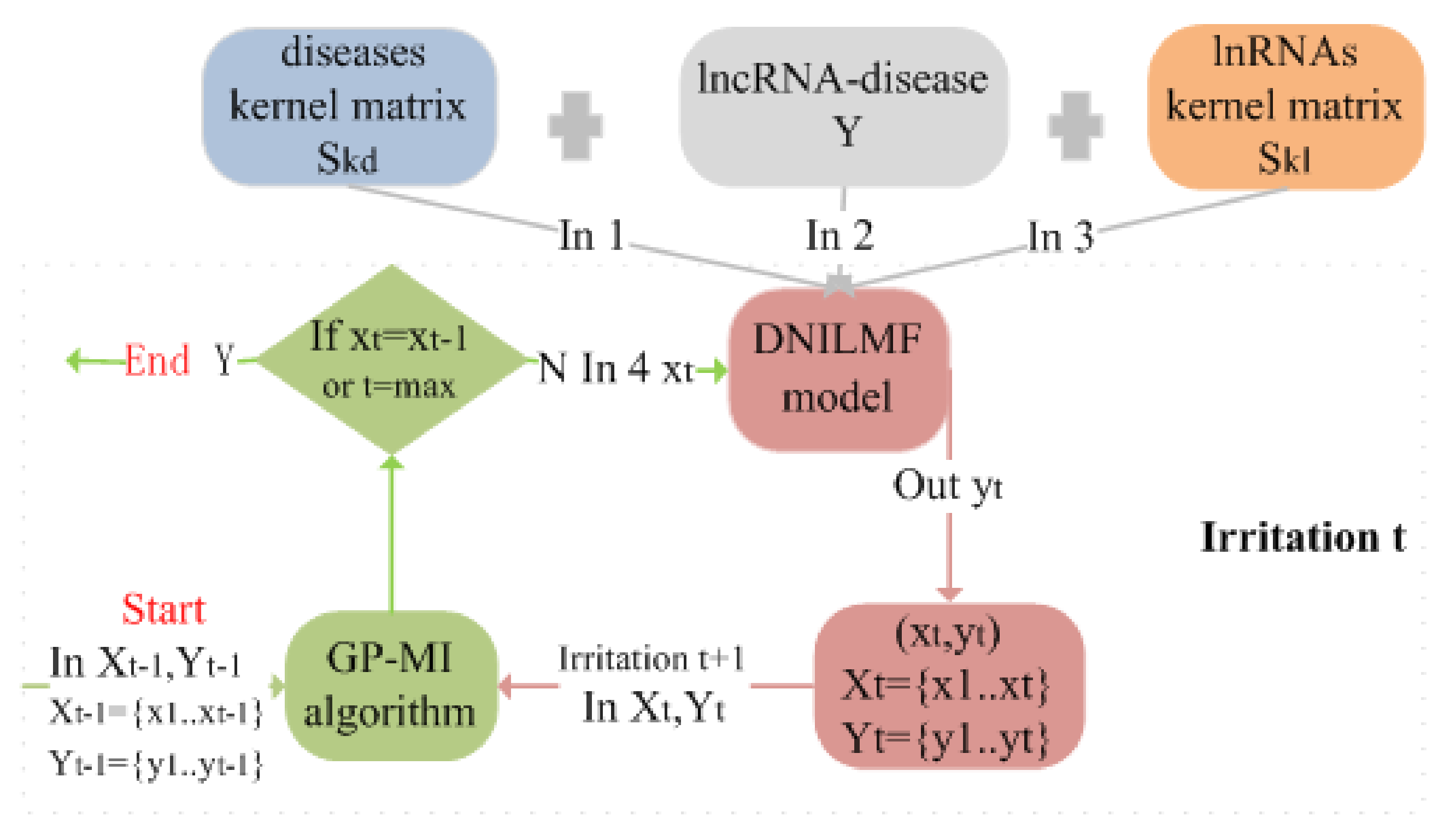
3.3. Bayesian Optimization
- (1)
- Bayesian optimization
- (2)
- Gaussian process
- (3)
- GP-MI algorithm
| Algorithm 1 GP-MI. |
fordo Compute and by and // Bayesian inference // Definition of for all // Selection of the next query location // Update get by the DNILMF model and // Query end for |
4. Experimental Results
4.1. Evaluation of Prediction Performance
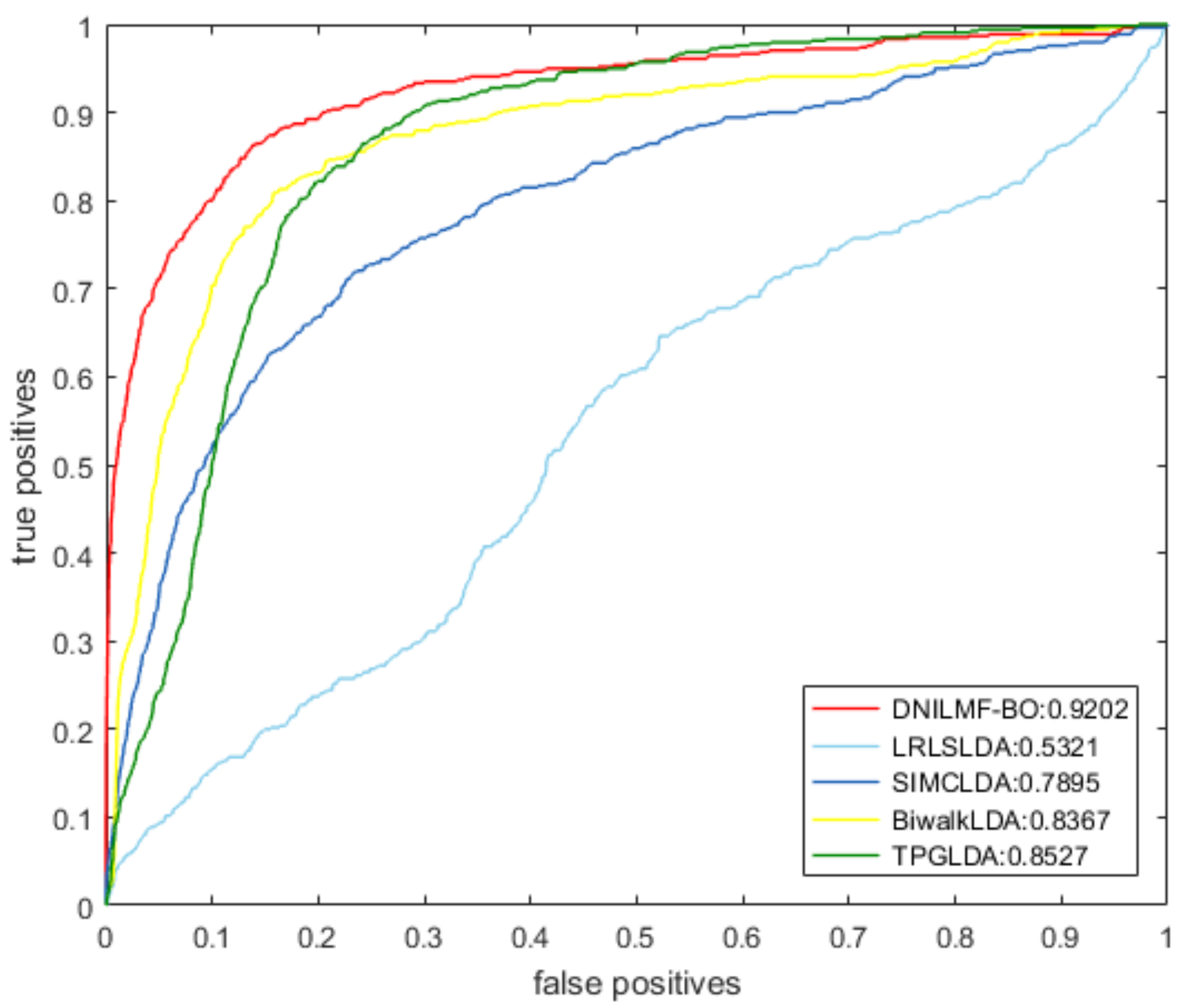
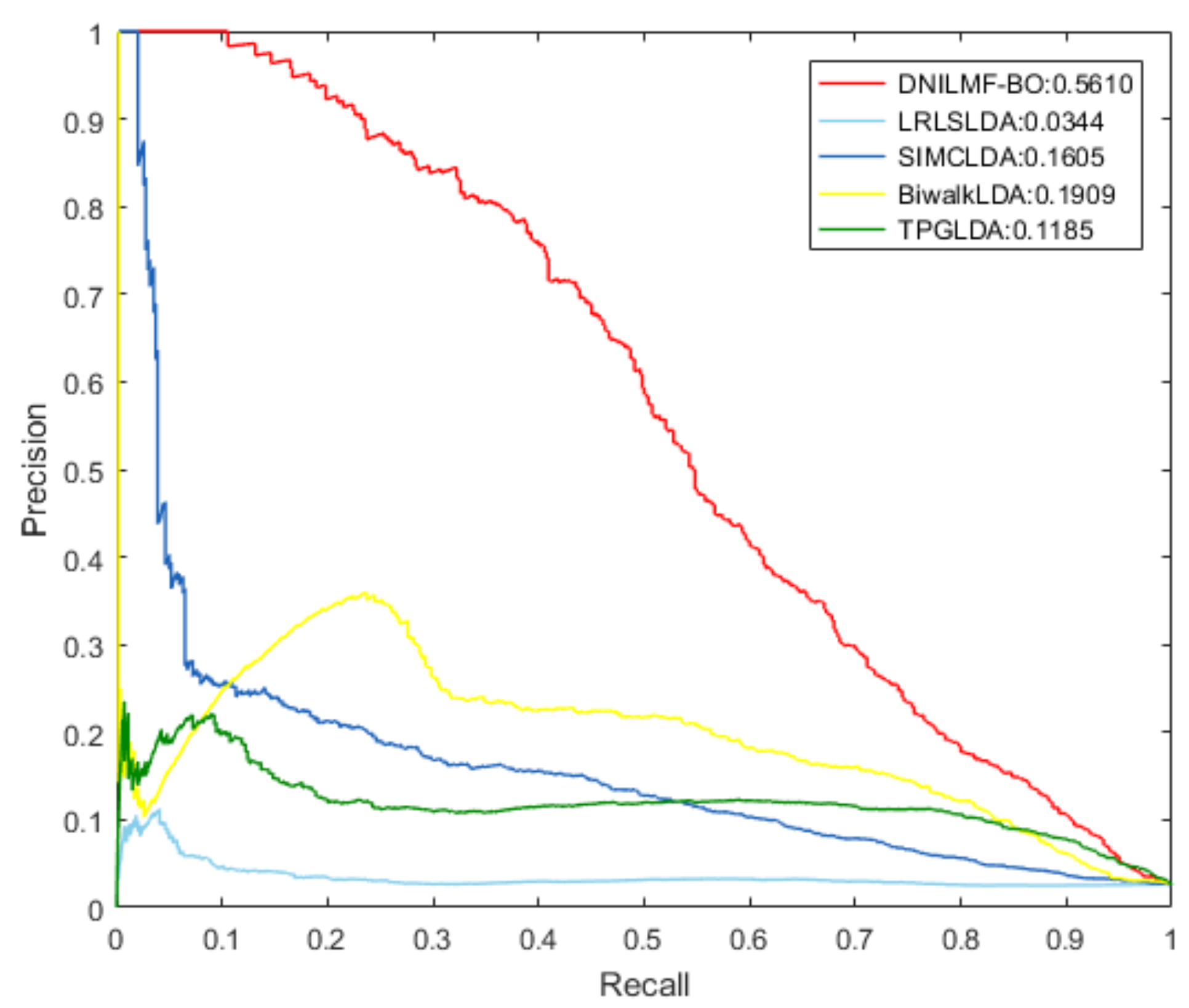
4.2. Comparison with Other Methods
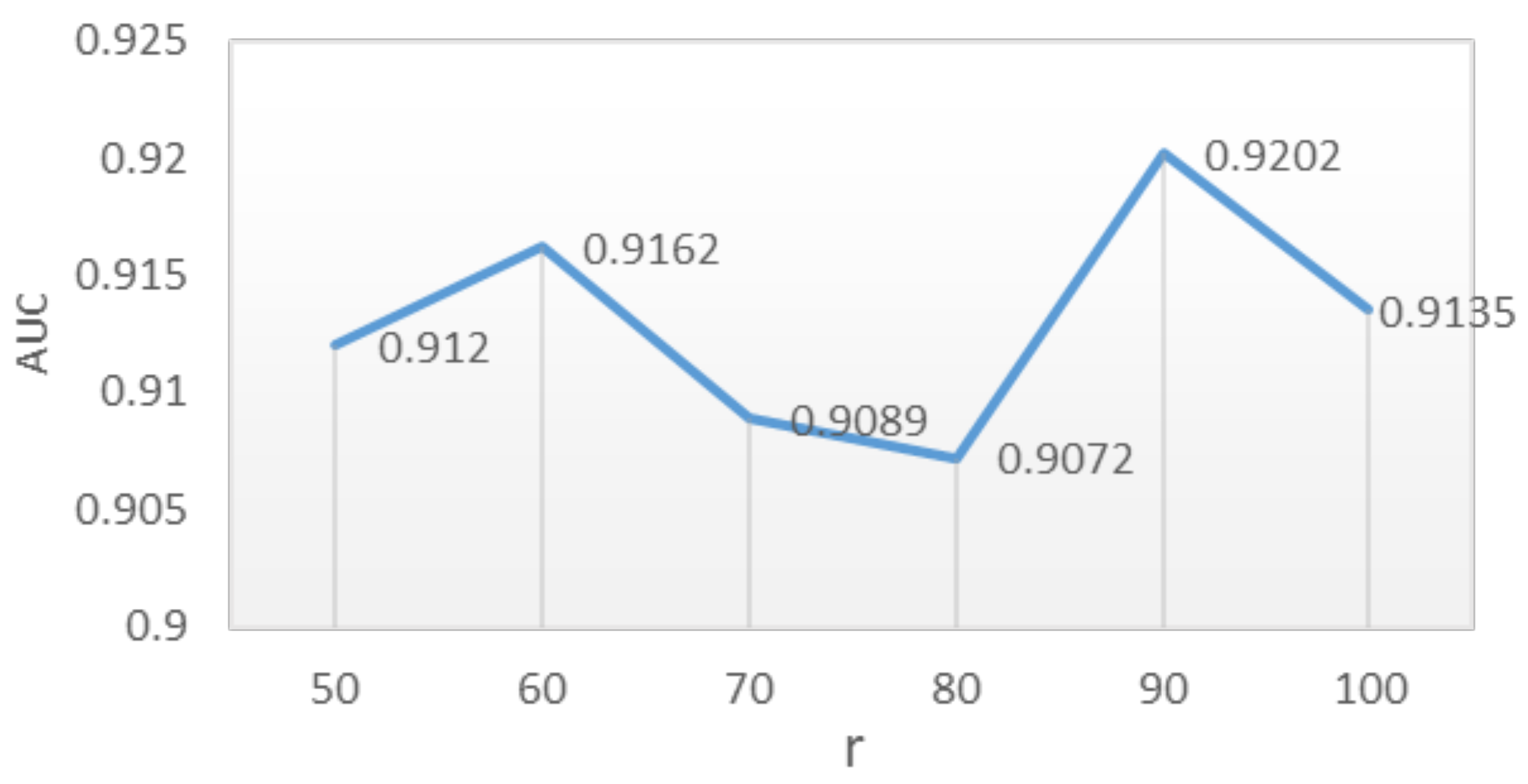
4.3. Parameter Analysis
4.4. Case Studies on Breast, Lung, and Colon Cancer
5. Discussion
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Chang, H.Y. Abstract IA02: Genome regulation by long noncoding RNAs. Cancer Res. 2016, 76, IA02. [Google Scholar]
- Yanofsky, C. Establishing the triplet nature of the genetic code. Cell 2007, 128, 815–818. [Google Scholar] [CrossRef] [PubMed]
- Merry, C.R.; Niland, C.; Khalil, A.M. Diverse Functions and Mechanisms of Mammalian Long Noncoding RNAs; Springer: New York, NY, USA, 2015; pp. 1–14. [Google Scholar]
- Cheetham, S.; Gruhl, F.; Mattick, J.; Dinger, M. Long noncoding RNAs and the genetics of cancer. Br. J. Cancer 2013, 108, 2419. [Google Scholar] [CrossRef] [PubMed]
- Taft, R.J.; Pang, K.C.; Mercer, T.R.; Dinger, M.; Mattick, J.S. Non-coding RNAs: Regulators of disease. J. Pathol. 2010, 220, 126–139. [Google Scholar] [CrossRef] [PubMed]
- Pasmant, E.; Sabbagh, A.; Vidaud, M.; Bièche, I. ANRIL, a long, noncoding RNA, is an unexpected major hotspot in GWAS. FASEB J. 2011, 25, 444–448. [Google Scholar] [CrossRef]
- Zhang, Q.; Chen, C.Y.; Yedavalli, V.S.; Jeang, K.T. NEAT1 long noncoding RNA and paraspeckle bodies modulate HIV-1 posttranscriptional expression. MBio 2013, 4, e00596–e00612. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Liu, X.; Wu, H.; Ni, P.; Gu, Z.; Qiao, Y.; Chen, N.; Sun, F.; Fan, Q. CREB up-regulates long non-coding RNA, HULC expression through interaction with microRNA-372 in liver cancer. Nucleic Acids Res. 2010, 38, 5366–5383. [Google Scholar] [CrossRef] [Green Version]
- Wapinski, O.; Chang, H.Y. Long noncoding RNAs and human disease. Trends Cell Biol. 2011, 21, 354–361. [Google Scholar] [CrossRef]
- Cui, Z.; Ren, S.; Lu, J.; Wang, F.; Xu, W.; Sun, Y.; Wei, M.; Chen, J.; Gao, X.; Xu, C.; et al. The prostate cancer-up-regulated long noncoding RNA PlncRNA-1 modulates apoptosis and proliferation through reciprocal regulation of androgen receptor. Urol. Oncol. Semin. Orig. Investig. 2013, 31, 1117–1123. [Google Scholar] [CrossRef]
- Sun, J.; Chen, X.; Wang, Z.; Guo, M.; Shi, H.; Wang, X.; Cheng, L.; Zhou, M. A potential prognostic long non-coding RNA signature to predict metastasis-free survival of breast cancer patients. Sci. Rep. 2015, 5, 16553. [Google Scholar] [CrossRef]
- Ma, Z.; Xue, S.; Zeng, B.; Qiu, D. lncRNA SNHG5 is associated with poor prognosis of bladder cancer and promotes bladder cancer cell proliferation through targeting p27. Trends Cell Biol. 2018, 15, 1924–1930. [Google Scholar] [CrossRef] [PubMed]
- Quek, X.C.; Thomson, D.W.; Maag, J.L.; Bartonicek, N.; Signal, B.; Clark, M.B.; Gloss, B.S.; Dinger, M.E. lncRNAdb v2. 0: Expanding the reference database for functional long noncoding RNAs. Nucleic Acids Res. 2014, 21, D168–D173. [Google Scholar]
- Chen, G.; Wang, Z.; Wang, D.; Qiu, C.; Liu, M.; Chen, X.; Zhang, Q.; Yan, G.; Cui, Q. lncRNADisease: A database for long-non-coding RNA-associated diseases. Nucleic Acids Res. 2012, 41, D983–D986. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Wang, P.; Wang, Y.; Ma, X.; Zhi, H.; Zhou, D.; Li, X.; Fang, Y.; Shen, W.; Xu, Y.; et al. Lnc2Cancer v2.0: Updated database of experimentally supported long non-coding RNAs in human cancers. Nucleic Acids Res. 2018, 47, D1028–D1033. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Li, H.; Fang, S.; Kang, Y.; Wu, W.; Hao, Y.; Li, Z.; Bu, D.; Sun, N.; Zhang, M.Q.; et al. NONCODE 2016: An informative and valuable data source of long non-coding RNAs. Nucleic Acids Res. 2015, 44, D203–D208. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Yan, G.Y. Novel human lncRNA-disease association inference based on lncRNA expression profiles. Bioinformatics 2013, 29, 2617–2624. [Google Scholar] [CrossRef]
- Lan, W.; Li, M.; Zhao, K.; Liu, J.; Wu, F.X.; Pan, Y.; Wang, J. LDAP: A web server for lncRNA-disease association prediction. Bioinformatics 2016, 33, 458–460. [Google Scholar] [CrossRef]
- Yu, J.; Ping, P.; Wang, L.; Kuang, L.; Li, X.; Wu, Z. A Novel Probability Model for lncRNA-Disease Association Prediction Based on the Naïve Bayesian Classifier. Genes 2018, 9, 345. [Google Scholar] [CrossRef]
- Ding, L.; Wang, M.; Sun, D.; Li, A. TPGLDA: Novel prediction of associations between lncRNAs and diseases via lncRNA-disease-gene tripartite graph. Sci. Rep. 2018, 8, 1065. [Google Scholar] [CrossRef]
- Chen, X.; You, Z.H.; Yan, G.Y.; Gong, D.W. IRWRLDA: Improved random walk with restart for lncRNA-disease association prediction. Oncotarget 2016, 7, 57919. [Google Scholar] [CrossRef]
- Gu, C.; Liao, B.; Li, X.; Cai, L.; Li, Z.; Li, K.; Yang, J. Global network random walk for predicting potential human lncRNA-disease associations. Sci. Rep. 2017, 7, 12442. [Google Scholar] [CrossRef]
- Fu, G.; Wang, J.; Domeniconi, C.; Yu, G. Matrix factorization-based data fusion for the prediction of lncRNA-disease associations. Bioinformatics 2017, 34, 1529–1537. [Google Scholar] [CrossRef]
- Lu, C.; Yang, M.; Luo, F.; Wu, F.X.; Li, M.; Pan, Y.; Li, Y.; Wang, J. Prediction of lncRNA-disease associations based on inductive matrix completion. Bioinformatics 2018, 34, 3357–3364. [Google Scholar] [CrossRef]
- Hao, M.; Bryant, S.H.; Wang, Y. Predicting drug–target interactions by dual-network integrated logistic matrix factorization. Sci. Rep. 2017, 7, 40376. [Google Scholar] [CrossRef]
- Liu, Y.; Wu, M.; Miao, C.; Zhao, P.; Li, X.L. Neighborhood regularized logistic matrix factorization for drug–target interaction prediction. PLoS Comput. Biol. 2016, 12, e1004760. [Google Scholar] [CrossRef]
- Yan, C.; Wang, J.; Ni, P.; Lan, W.; Wu, F.; Pan, Y. DNRLMF-MDA: Predicting microRNA-disease associations based on similarities of microRNAs and diseases. IEEE/ACM Trans. Comput. Biol. Bioinform. 2017, 16, 233–243. [Google Scholar] [CrossRef]
- Zhao, Q.; Zhang, Y.; Hu, H.; Ren, G.; Zhang, W.; Liu, H. IRWNRLPI: Integrating random walk and neighborhood regularized logistic matrix factorization for lncRNA-protein interaction prediction. Front. Genet. 2018, 9, 239. [Google Scholar] [CrossRef]
- Liu, H.; Ren, G.; Hu, H.; Zhang, L.; Ai, H.; Zhang, W.; Zhao, Q. LPI-NRLMF: lncRNA-protein interaction prediction by neighborhood regularized logistic matrix factorization. Oncotarget 2017, 8, 103975. [Google Scholar] [CrossRef]
- Contal, E.; Perchet, V.; Vayatis, N. Gaussian process optimization with mutual information. In Proceedings of the International Conference on Machine Learning, Beijing, China, 21–26 June 2014; pp. 253–261. [Google Scholar]
- Ban, T.; Ohue, M.; Akiyama, Y. Efficient hyperparameter optimization by using Bayesian optimization for drug–target interaction prediction. In Proceedings of the 2017 IEEE 7th International Conference on Computational Advances in Bio and Medical Sciences (ICCABS), Orlando, FL, USA, 19–21 October 2017; pp. 1–6. [Google Scholar]
- Parkinson, H.; Kapushesky, M.; Shojatalab, M.; Abeygunawardena, N.; Coulson, R.; Farne, A.; Holloway, E.; Kolesnykov, N.; Lilja, P.; Lukk, M.; et al. ArrayExpress—A public database of microarray experiments and gene expression profiles. Nucleic Acids Res. 2006, 35, D747–D750. [Google Scholar] [CrossRef]
- Wang, D.; Wang, J.; Lu, M.; Song, F.; Cui, Q. Inferring the human microRNA functional similarity and functional network based on microRNA-associated diseases. Bioinformatics 2010, 26, 1644–1650. [Google Scholar] [CrossRef] [Green Version]
- Yu, G.; Wang, L.G.; Yan, G.R.; He, Q.Y. DOSE: An R/Bioconductor package for disease ontology semantic and enrichment analysis. Bioinformatics 2014, 31, 608–609. [Google Scholar] [CrossRef]
- Van Laarhoven, T.; Nabuurs, S.B.; Marchiori, E. Gaussian interaction profile kernels for predicting drug–target interaction. Bioinformatics 2011, 27, 3036–3043. [Google Scholar] [CrossRef]
- Hao, M.; Wang, Y.; Bryant, S.H. Improved prediction of drug–target interactions using regularized least squares integrating with kernel fusion technique. Anal. Chim. Acta 2016, 909, 41–50. [Google Scholar] [CrossRef]
- Wang, B.; Mezlini, A.M.; Demir, F.; Fiume, M.; Tu, Z.; Brudno, M.; Haibe-Kains, B.; Goldenberg, A. Similarity network fusion for aggregating data types on a genomic scale. Nat. Methods 2014, 11, 333. [Google Scholar] [CrossRef]
- Duchi, J.; Hazan, E.; Singer, Y. Adaptive subgradient methods for online learning and stochastic optimization. J. Mach. Learn. Res. 2011, 12, 2121–2159. [Google Scholar]
- Nie, F.Q.; Sun, M.; Yang, J.S.; Xie, M.; Xu, T.P.; Xia, R.; Liu, Y.W.; Liu, X.H.; Zhang, E.B.; Lu, K.H.; et al. Long noncoding RNA ANRIL promotes non-small cell lung cancer cell proliferation and inhibits apoptosis by silencing KLF2 and P21 expression. Mol. Cancer Ther. 2015, 14, 268–277. [Google Scholar] [CrossRef]
- Wang, H.M.; Lu, J.H.; Chen, W.Y.; Gu, A.Q. Upregulated lncRNA-UCA1 contributes to progression of lung cancer and is closely related to clinical diagnosis as a predictive biomarker in plasma. Int. J. Clin. Exp. Med. 2015, 8, 11824. [Google Scholar]
- Prenner, S.; Levitsky, J. Comprehensive review on colorectal cancer and transplant. Am. J. Transplant. 2017, 17, 2761–2774. [Google Scholar] [CrossRef]
- Zhai, H.Y.; Sui, M.H.; Yu, X.; Qu, Z.; Hu, J.C.; Sun, H.Q.; Zheng, H.T.; Zhou, K.; Jiang, L.X. Overexpression of long non-coding RNA TUG1 promotes colon cancer progression. Med. Sci. Monit. 2016, 22, 3281. [Google Scholar] [CrossRef]
- Zhang, X.F.; Liu, T.; Li, Y.; Li, S. Overexpression of long non-coding RNA CCAT1 is a novel biomarker of poor prognosis in patients with breast cancer. Int. J. Clin. Exp. Pathol. 2015, 8, 9440. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Method | AUC | AUPR |
|---|---|---|
| LRLSLDA | 0.5321 | 0.0344 |
| SIMCLDA | 0.7895 | 0.1605 |
| BiwalkLDA | 0.8367 | 0.1909 |
| TPGLDA | 0.8527 | 0.1185 |
| DNILMF-LDA | 0.9202 | 0.5610 |
| Top | lncRNA | Evidence | Description |
|---|---|---|---|
| 1 | CCAT2 | 26729200 | lncRNADisease |
| 2 | CDKN2B-AS1 | 26729200 | lncRNADisease |
| 3 | PVT1 | 28731781 | lnc2Cancer |
| 4 | UCA1 | 29731641 | lnc2Cancer |
| 5 | CCAT1 | 27212446 | lncRNADisease |
| 6 | SPRY4-IT1 | 26302345 | lncRNADisease |
| 7 | GAS5 | 26634743 | lncRNADisease |
| 8 | HULC | unconfirmed | unconfirmed |
| 9 | SRA1 | unconfirmed | unconfirmed |
| 10 | XIST | unconfirmed | unconfirmed |
| Top | lncRNA | Evidence | Description |
|---|---|---|---|
| 1 | SPRY4-IT1 | 28099409 | lnc2Cancer |
| 2 | HOTTIP | 26617875 | lnc2Cancer |
| 3 | GHET1 | 27931286 | lnc2Cancer |
| 4 | MINA | unconfirmed | unconfirmed |
| 5 | HIF1A-AS2 | 29278853 | lnc2Cancer |
| 6 | ADAMTS9-AS2 | 27596298 | lnc2Cancer |
| 7 | TUG1 | 28302487 | lnc2Cancer |
| 8 | LINC00152 | 29180678 | lnc2Cancer |
| 9 | PANDAR | 28176943 | lnc2Cancer |
| 10 | BC040587 | unconfirmed | unconfirmed |
| Top | lncRNA | Evidence | Description |
|---|---|---|---|
| 1 | MNX1-AS1 | unconfirmed | unconfirmed |
| 2 | CCAT1 | 26464701 | lnc2Cancer |
| 3 | TUSC7 | 23558749 | lnc2Cancer |
| 4 | BANCR | 29565494 | lnc2Cancer |
| 5 | DNM3OS | unconfirmed | unconfirmed |
| 6 | TUG1 | 27791993 | lncRNADisease |
| 7 | RPL34-AS1 | unconfirmed | lncRNADisease |
| 8 | MINA | 25586347 | lnc2Cancer |
| 9 | GHET1 | 29843220 | lnc2Cancer |
| 10 | PTENP1 | 29085464 | lnc2Cancer |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Y.; Li, J.; Bian, N. DNILMF-LDA: Prediction of lncRNA-Disease Associations by Dual-Network Integrated Logistic Matrix Factorization and Bayesian Optimization. Genes 2019, 10, 608. https://doi.org/10.3390/genes10080608
Li Y, Li J, Bian N. DNILMF-LDA: Prediction of lncRNA-Disease Associations by Dual-Network Integrated Logistic Matrix Factorization and Bayesian Optimization. Genes. 2019; 10(8):608. https://doi.org/10.3390/genes10080608
Chicago/Turabian StyleLi, Yan, Junyi Li, and Naizheng Bian. 2019. "DNILMF-LDA: Prediction of lncRNA-Disease Associations by Dual-Network Integrated Logistic Matrix Factorization and Bayesian Optimization" Genes 10, no. 8: 608. https://doi.org/10.3390/genes10080608