Transcriptome Changes during Major Developmental Transitions Accompanied with Little Alteration of DNA Methylome in Two Pleurotus Species

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Strains and Culture Conditions
2.2. Whole-Genome Bisulfite Sequencing and Data Processing
2.3. Differential Methylation Analysis
2.4. RNA-seq and Data Process
2.5. Gene Ontology (GO) Enrichment Analysis
2.6. Statistics
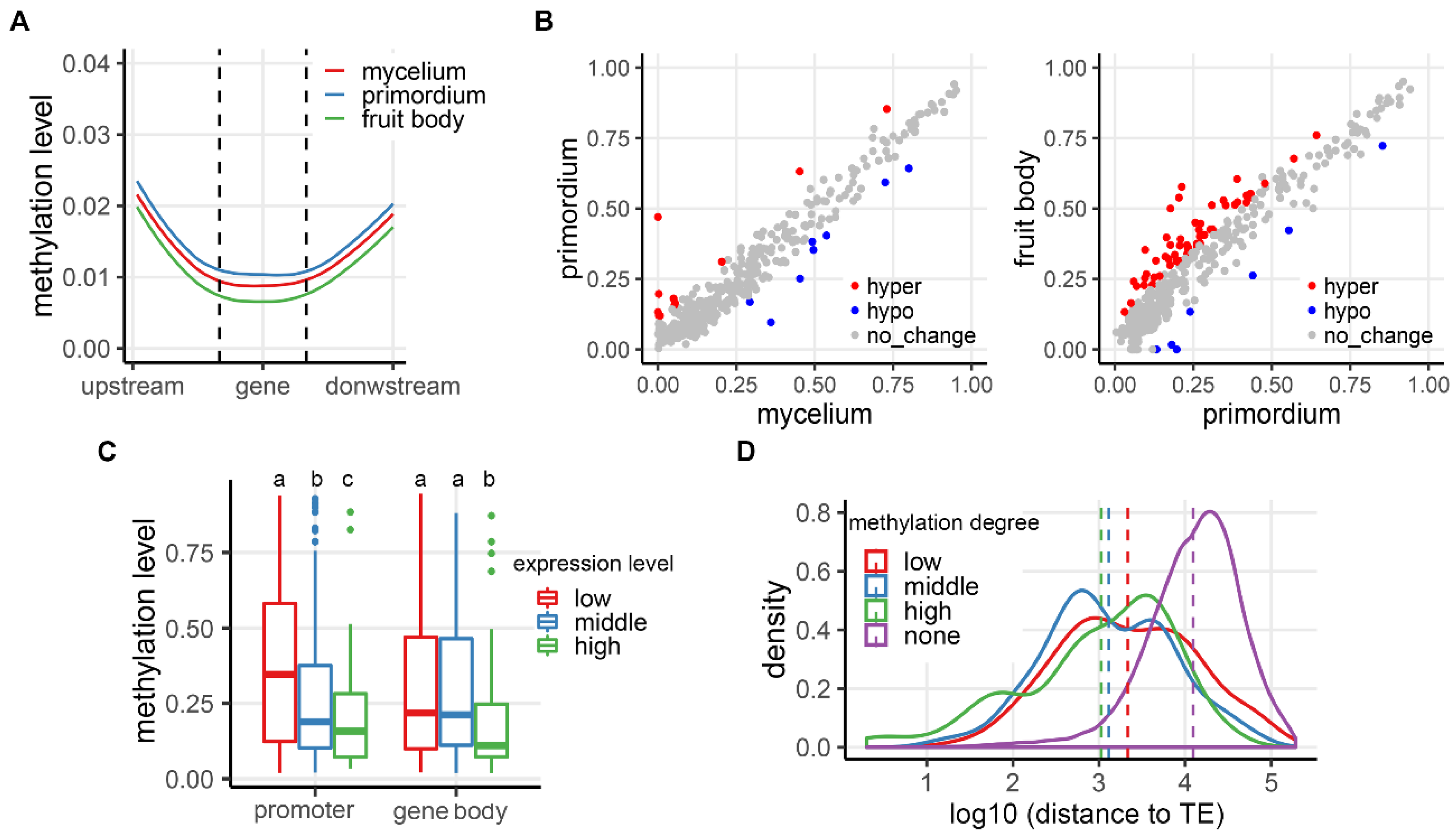
3. Results
3.1. DNA Methylation Landscapes in the Two Mushroom Species
3.2. Transcriptional Regulation of Gene Expression in the Two Mushroom Species
3.3. Correlation Between CG Methylation and Expression of TEs during Development in the Two Mushroom Species
3.4. Correlation of CG Methylation and Expression in Protein-Coding Genes
4. Discussion
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Zervakis, G.I.; Venturella, G.; Papadopoulou, K. Genetic polymorphism and taxonomic infrastructure of the Pleurotus eryngii species-complex as determined by RAPD analysis, isozyme profiles and ecomorphological characters. Microbiology 2001, 147, 3183–3194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tolgor, L. The species resource and ecological distribution of Pleurotus in China. Edible Fungi China 2001, 20, 8–10. [Google Scholar]
- Zervakis, G.I.; Ntougias, S.; Gargano, M.L.; Besi, M.I.; Polemis, E.; Typas, M.A.; Venturella, G. A reappraisal of the Pleurotus eryngii complex—New species and taxonomic combinations based on the application of a polyphasic approach, and an identification key to Pleurotus taxa associated with Apiaceae plants. Fungal Biol. 2014, 118, 814–834. [Google Scholar] [CrossRef] [PubMed]
- Kawai, G.; Babasaki, K.; Neda, H. Taxonomic position of a Chinese Pleurotus “Bai-Ling-Gu”: It belongs to Pleurotus eryngii (DC.: Fr.) Quél. and evolved independently in China. Mycoscience 2008, 49, 75–87. [Google Scholar] [CrossRef]
- Kim, M.K.; Ryu, J.-S.; Lee, Y.-H.; Kim, H.-R. Breeding of a long shelf-life strain for commercial cultivation by mono–mono crossing in Pleurotus eryngii. Sci. Hortic. 2013, 162, 265–270. [Google Scholar] [CrossRef]
- Kües, U.; Liu, Y. Fruiting body production in basidiomycetes. Appl. Microbiol. Biotechnol. 2000, 54, 141–152. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.P.; Dai, Y.T.; Yang, C.T.; Wei, P.; Song, B.; Yang, Y.; Sun, L.; Zhang, Z.W.; Li, Y. Comparative Transcriptome Analysis Identified Candidate Genes Related to Bailinggu Mushroom Formation and Genetic Markers for Genetic Analyses and Breeding. Sci. Rep. 2017, 7, 9266. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Wen, J.; Li, J.; Ma, X.; Yu, Y.; Tan, X.; Wang, Q.; Liu, B.; Li, X.; Li, Y. The evolution of genomic and epigenomic features in two Pleurotus fungi. Sci. Rep. 2018, 8, 8313. [Google Scholar] [CrossRef] [PubMed]
- Martienssen, R.A.; Colot, V. DNA methylation and epigenetic inheritance in plants and filamentous fungi. Science 2001, 293, 1070. [Google Scholar] [CrossRef] [PubMed]
- Becker, C.; Hagmann, J.; Müller, J.; Koenig, D.; Stegle, O.; Borgwardt, K.; Weigel, D. Spontaneous epigenetic variation in the Arabidopsis thaliana methylome. Nature 2011, 480, 245. [Google Scholar] [CrossRef] [PubMed]
- Lister, R.; Pelizzola, M.; Dowen, R.H.; Hawkins, R.D.; Hon, G.; Tonti-Filippini, J.; Nery, J.R.; Lee, L.; Ye, Z.; Ngo, Q.-M. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature 2009, 462, 315. [Google Scholar] [CrossRef] [PubMed]
- Law, J.A.; Jacobsen, S.E. Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nat. Rev. Genet. 2010, 11, 204–220. [Google Scholar] [CrossRef] [PubMed]
- Smith, Z.D.; Meissner, A. DNA methylation: Roles in mammalian development. Nat. Rev. Genet. 2013, 14, 204–220. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Liu, R.; Niu, Q.; Tang, K.; Zhang, B.; Zhang, H.; Chen, K.; Zhu, J.-K.; Lang, Z. Global increase in DNA methylation during orange fruit development and ripening. Proc. Natl. Acad. Sci. USA 2019, 116, 1430–1436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cokus, S.J.; Feng, S.; Zhang, X.; Chen, Z.; Merriman, B.; Haudenschild, C.D.; Pradhan, S.; Nelson, S.F.; Pellegrini, M.; Jacobsen, S.E. Shotgun bisulphite sequencing of the Arabidopsis genome reveals DNA methylation patterning. Nature 2008, 452, 215. [Google Scholar] [CrossRef] [PubMed]
- Ziller, M.J.; Gu, H.; Müller, F.; Donaghey, J.; Tsai, L.T.-Y.; Kohlbacher, O.; De Jager, P.L.; Rosen, E.D.; Bennett, D.A.; Bernstein, B.E. Charting a dynamic DNA methylation landscape of the human genome. Nature 2013, 500, 477. [Google Scholar] [CrossRef] [PubMed]
- Montanini, B.; Chen, P.Y.; Morselli, M.; Jaroszewicz, A.; Lopez, D.; Martin, F.; Ottonello, S.; Pellegrini, M. Non-exhaustive DNA methylation-mediated transposon silencing in the black truffle genome, a complex fungal genome with massive repeat element content. Genome Biol. 2014, 15, 411. [Google Scholar] [CrossRef] [PubMed]
- Zemach, A.; McDaniel, I.E.; Silva, P.; Zilberman, D. Genome-wide evolutionary analysis of eukaryotic DNA methylation. Science 2010, 328, 916–919. [Google Scholar] [CrossRef] [PubMed]
- Castanera, R.; Lopez-Varas, L.; Borgognone, A.; LaButti, K.; Lapidus, A.; Schmutz, J.; Grimwood, J.; Perez, G.; Pisabarro, A.G.; Grigoriev, I.V.; et al. Transposable Elements versus the Fungal Genome: Impact on Whole-Genome Architecture and Transcriptional Profiles. PLoS Genet. 2016, 12, e1006108. [Google Scholar] [CrossRef] [PubMed]
- Borgognone, A.; Castanera, R.; Morselli, M.; López-Varas, L.; Rubbi, L.; Pisabarro, A.G.; Pellegrini, M.; Ramírez, L. Transposon-associated epigenetic silencing during Pleurotus ostreatus life cycle. DNA Res. 2018, 25, 451–464. [Google Scholar] [CrossRef]
- Seisenberger, S.; Andrews, S.; Krueger, F.; Arand, J.; Walter, J.; Santos, F.; Popp, C.; Thienpont, B.; Dean, W.; Reik, W. The dynamics of genome-wide DNA methylation reprogramming in mouse primordial germ cells. Mol. Cell 2012, 48, 849–862. [Google Scholar] [CrossRef] [PubMed]
- Smith, Z.D.; Chan, M.M.; Mikkelsen, T.S.; Gu, H.; Gnirke, A.; Regev, A.; Meissner, A. A unique regulatory phase of DNA methylation in the early mammalian embryo. Nature 2012, 484, 339. [Google Scholar] [CrossRef] [PubMed]
- Zemach, A.; Kim, M.Y.; Silva, P.; Rodrigues, J.A.; Dotson, B.; Brooks, M.D.; Zilberman, D. Local DNA hypomethylation activates genes in rice endosperm. Proc. Natl. Acad. Sci. USA 2010, 107, 18729–18734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, S.; Fei, Z.; Chen, Y.-R.; Zheng, Y.; Huang, M.; Vrebalov, J.; McQuinn, R.; Gapper, N.; Liu, B.; Xiang, J. Single-base resolution methylomes of tomato fruit development reveal epigenome modifications associated with ripening. Nat. Biotechnol. 2013, 31, 154. [Google Scholar] [CrossRef] [PubMed]
- Daccord, N.; Celton, J.-M.; Linsmith, G.; Becker, C.; Choisne, N.; Schijlen, E.; van de Geest, H.; Bianco, L.; Micheletti, D.; Velasco, R. High-quality de novo assembly of the apple genome and methylome dynamics of early fruit development. Nat. Genet. 2017, 49, 1099. [Google Scholar] [CrossRef] [PubMed]
- Jeon, J.; Choi, J.; Lee, G.W.; Park, S.Y.; Huh, A.; Dean, R.A.; Lee, Y.H. Genome-wide profiling of DNA methylation provides insights into epigenetic regulation of fungal development in a plant pathogenic fungus, Magnaporthe oryzae. Sci. Rep. 2015, 5, 8567. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-L.; Wang, Z.-X.; Liu, C.; Wang, S.-B.; Huang, B. Genome-wide analysis of DNA methylation in the sexual stage of the insect pathogenic fungus Cordyceps militaris. Fungal Biol. 2015, 119, 1246–1254. [Google Scholar] [CrossRef]
- Borin, G.P.; Sanchez, C.C.; Santana, E.S.; Zanini, G.K.; Santos, R.A.C.; Pontes, A.O.; Souza, A.T.; Dal, R.M.M.T.S.; Riaño-Pachón, D.M.; Goldman, G.H. Comparative transcriptome analysis reveals different strategies for degradation of steam-exploded sugarcane bagasse by Aspergillus niger and Trichoderma reesei. BMC Genom. 2017, 18, 501. [Google Scholar] [CrossRef] [PubMed]
- Qiu, D.; Xu, L.; Vandemark, G.; Chen, W. Comparative transcriptome analysis between the fungal plant pathogens Sclerotinia sclerotiorum and S. trifoliorum using RNA sequencing. J. Hered. 2015, 107, 163–172. [Google Scholar] [CrossRef]
- Plaza, D.F.; Lin, C.-W.; van der Velden, N.S.J.; Aebi, M.; Künzler, M. Comparative transcriptomics of the model mushroom Coprinopsis cinerea reveals tissue-specific armories and a conserved circuitry for sexual development. BMC Genom. 2014, 15, 492. [Google Scholar] [CrossRef]
- Hu, L.; Li, N.; Xu, C.; Zhong, S.; Lin, X.; Yang, J.; Zhou, T.; Yuliang, A.; Wu, Y.; Chen, Y.-R. Mutation of a major CG methylase in rice causes genome-wide hypomethylation, dysregulated genome expression, and seedling lethality. Proc. Natl. Acad. Sci. USA 2014, 111, 10642–10647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114. [Google Scholar] [CrossRef] [PubMed]
- Krueger, F.; Andrews, S.R. Bismark: A flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics 2011, 27, 1571–1572. [Google Scholar] [CrossRef] [PubMed]
- Schultz, M.D.; Schmitz, R.J.; Ecker, J.R. ‘Leveling’ the playing field for analyses of single-base resolution DNA methylomes. Trends Genet. 2012, 28, 583–585. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Aageaard, A.; Bechsgaard, J.; Bilde, T. DNA Methylation Patterns in the Social Spider, Stegodyphus dumicola. Genes 2019, 10, 137. [Google Scholar] [CrossRef] [PubMed]
- Pertea, M.; Kim, D.; Pertea, G.; Leek, J.T.; Salzberg, S.L. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie, and Ballgown. Nat. Protoc. 2016, 11, 1650. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Stoeckert, C.J.; Roos, D.S. OrthoMCL: Identification of Ortholog Groups for Eukaryotic Genomes. Genome Res. 2003, 13, 2178. [Google Scholar] [CrossRef] [PubMed]
- Kent, W.J. BLAT—The BLAST-like alignment tool. Genome Res. 2002, 12, 656–664. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. clusterProfiler: An R Package for Comparing Biological Themes among Gene Clusters. Omics J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Zhang, Y.; Ngu, D.W.; Carvalho, D.; Liang, Z.; Qiu, Y.; Roston, R.L.; Schnable, J.C. Differentially regulated orthologs in sorghum and the subgenomes of maize. Plant Cell 2017, 29, 1938–1951. [Google Scholar] [CrossRef] [PubMed]
- Heinfling, A.; Martínez, M.J.; Martínez, A.T.; Bergbauer, M.; Szewzyk, U. Transformation of industrial dyes by manganese peroxidases from Bjerkandera adusta and Pleurotus eryngii in a manganese-independent reaction. Appl. Environ. Microbiol. 1998, 64, 2788–2793. [Google Scholar] [PubMed]
- Kwak, W.S.; Jung, S.H.; Kim, Y.I. Broiler litter supplementation improves storage and feed-nutritional value of sawdust-based spent mushroom substrate. Bioresour. Technol. 2008, 99, 2947–2955. [Google Scholar] [CrossRef] [PubMed]
- Nuhu, A.; Ki, N.Y.; Tae, S.L. Evaluation of the antioxidant and antityrosinase activities of three extracts from Pleurotus nebrodensis fruiting bodies. Afr. J. Biotechnol. 2011, 10, 2978–2986. [Google Scholar] [CrossRef]
- Estrada, A.E.; del Mar Jimenez-Gasco, M.; Royse, D.J. Pleurotus eryngii species complex: Sequence analysis and phylogeny based on partial EF1alpha and RPB2 genes. Fungal Biol. 2010, 114, 421–428. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Srivilai, P.; Loos, S.; Aebi, M.; Kües, U. An essential gene for fruiting body initiation in the basidiomycete Coprinopsis cinerea is homologous to bacterial cyclopropane fatty acid synthase genes. Genetics 2006, 172, 873–884. [Google Scholar] [CrossRef] [PubMed]
- Muraguchi, H.; Umezawa, K.; Niikura, M.; Yoshida, M.; Kozaki, T.; Ishii, K.; Sakai, K.; Shimizu, M.; Nakahori, K.; Sakamoto, Y. Strand-specific RNA-seq analyses of fruiting body development in Coprinopsis cinerea. PLoS ONE 2015, 10, e0141586. [Google Scholar] [CrossRef] [PubMed]
- Sikhakolli, U.R.; López-Giráldez, F.; Li, N.; Common, R.; Townsend, J.P.; Trail, F. Transcriptome analyses during fruiting body formation in Fusarium graminearum and Fusarium verticillioides reflect species life history and ecology. Fungal Genet. Biol. 2012, 49, 663–673. [Google Scholar] [CrossRef]
- Wang, M.; Gu, B.; Huang, J.; Jiang, S.; Chen, Y.; Yin, Y.; Pan, Y.; Yu, G.; Li, Y.; Wong, B.H.C. Transcriptome and proteome exploration to provide a resource for the study of Agrocybe aegerita. PLoS ONE 2013, 8, e56686. [Google Scholar] [CrossRef]
- Song, H.-Y.; Kim, D.-H.; Kim, J.-M. Comparative transcriptome analysis of dikaryotic mycelia and mature fruiting bodies in the edible mushroom Lentinula edodes. Sci. Rep. 2018, 8, 8983. [Google Scholar] [CrossRef]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wen, J.; Zhang, Z.; Gong, L.; Xun, H.; Li, J.; Qi, B.; Wang, Q.; Li, X.; Li, Y.; Liu, B. Transcriptome Changes during Major Developmental Transitions Accompanied with Little Alteration of DNA Methylome in Two Pleurotus Species. Genes 2019, 10, 465. https://doi.org/10.3390/genes10060465
Wen J, Zhang Z, Gong L, Xun H, Li J, Qi B, Wang Q, Li X, Li Y, Liu B. Transcriptome Changes during Major Developmental Transitions Accompanied with Little Alteration of DNA Methylome in Two Pleurotus Species. Genes. 2019; 10(6):465. https://doi.org/10.3390/genes10060465
Chicago/Turabian StyleWen, Jiawei, Zhibin Zhang, Lei Gong, Hongwei Xun, Juzuo Li, Bao Qi, Qi Wang, Xiaomeng Li, Yu Li, and Bao Liu. 2019. "Transcriptome Changes during Major Developmental Transitions Accompanied with Little Alteration of DNA Methylome in Two Pleurotus Species" Genes 10, no. 6: 465. https://doi.org/10.3390/genes10060465