Sequencing and Phylogenetic Analysis of Chloroplast Genes in Freshwater Raphidophytes


Abstract
:1. Introduction
2. Materials and Methods
3. Results
3.1. Processing of Reads
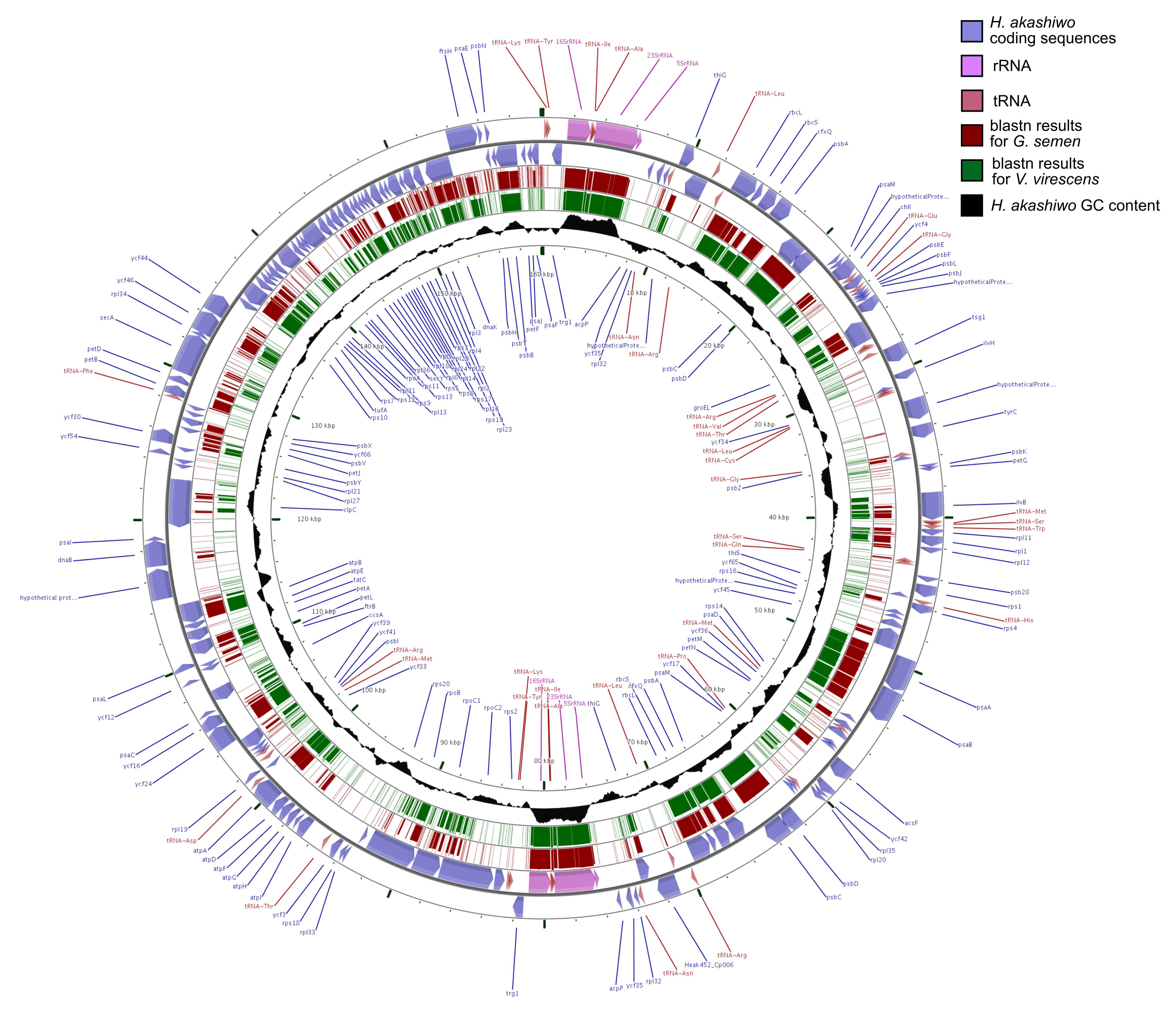
3.2. Annotation
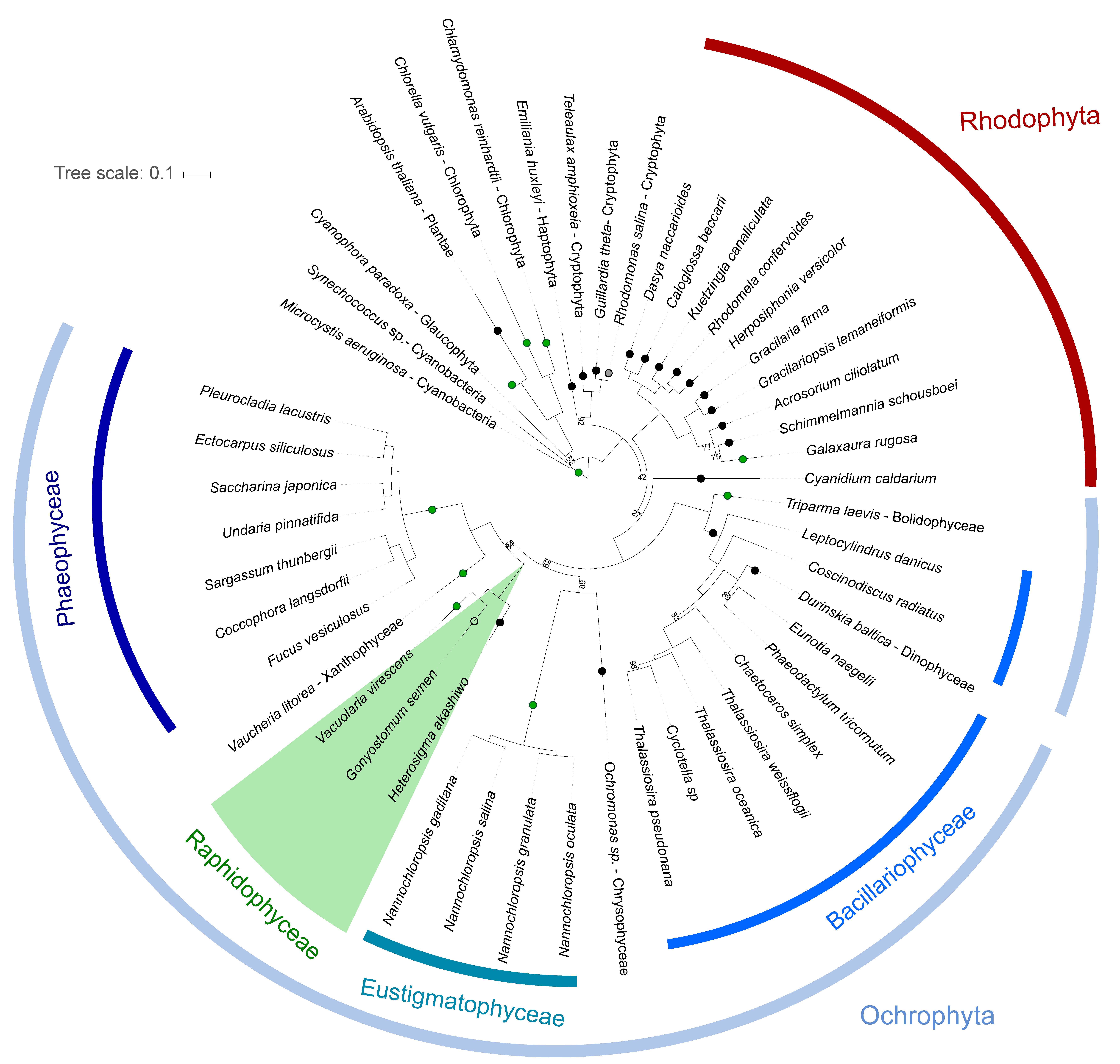
3.3. Phylogenetic Analyses
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Archibald, J.M. The Puzzle of Plastid Evolution. Curr. Biol. 2009, 19, R81–R88. [Google Scholar] [PubMed]
- Keeling, P.J. The endosymbiotic origin, diversification and fate of plastids. R. Soc. Philos. Trans. Biol. Sci. 2010, 365, 729–748. [Google Scholar] [CrossRef] [PubMed]
- Horiguchi, T. Raphidophyceae (Raphidophyta). In Handbook of the Protists; Archibald, J.M., Simpson, A.G.B., Slamovits, C.H., Margulis, L., Melkonian, M., Chapman, D.J., Corliss, J.O., Eds.; Springer International Publishing: Cham, Switzerland, 2016; pp. 1–26. ISBN 978-3-319-32669-6. [Google Scholar]
- Sassenhagen, I.; Rengefors, K.; Richardson, T.L.; Pinckney, J.L. Pigment composition and photoacclimation as keys to the ecological success of Gonyostomum semen (Raphidophyceae, Stramenopiles). J. Phycol. 2014, 50, 1146–1154. [Google Scholar] [CrossRef] [PubMed]
- Mostaert, A.S.; Karsten, U.; Hara, Y.; Watanabe, M.M. Pigments and fatty acids of marine raphidophytes: A chemotaxonomic re-evaluation. Phycol. Res. 1998, 46, 213–220. [Google Scholar] [CrossRef]
- Mason, C.B.; Matthews, S.; Bricker, T.M.; Moroney, J.V. Simplified Procedure for the Isolation of Intact Chloroplasts from Chlamydomonas reinhardtii. Plant Physiol. 1991, 97, 1576–1580. [Google Scholar] [PubMed]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 1 May 2016).
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Leung, H.C.M.; Yiu, S.M.; Chin, F.Y.L. IDBA-UD: A de novo assembler for single-cell and metagenomic sequencing data with highly uneven depth. Bioinformatics 2012, 28, 1420–1428. [Google Scholar] [CrossRef]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A New Genome Assembly Algorithm and Its Applications to Single-Cell Sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef]
- Nurk, S.; Meleshko, D.; Korobeynikov, A.; Pevzner, P.A. metaSPAdes: A new versatile metagenomic assembler. Genome Res. 2017, 27, 824–834. [Google Scholar] [CrossRef]
- Gurevich, A.; Saveliev, V.; Vyahhi, N.; Tesler, G. QUAST: Quality assessment tool for genome assemblies. Bioinforma. Oxf. Engl. 2013, 29, 1072–1075. [Google Scholar] [CrossRef]
- Eren, A.M.; Esen, Ö.C.; Quince, C.; Vineis, J.H.; Morrison, H.G.; Sogin, M.L.; Delmont, T.O. Anvi’o: An advanced analysis and visualization platform for ‘omics data. PeerJ 2015, 3, e1319. [Google Scholar]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: architecture and applications. BMC Bioinformatics 2009, 10, 421. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar]
- Albertsen, M.; Hugenholtz, P.; Skarshewski, A.; Nielsen, K.L.; Tyson, G.W.; Nielsen, P.H. Genome sequences of rare, uncultured bacteria obtained by differential coverage binning of multiple metagenomes. Nat. Biotechnol. 2013, 31, 533–538. [Google Scholar] [CrossRef]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10, R25. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Darling, A.E.; Jospin, G.; Lowe, E.; Matsen, F.A.; Bik, H.M.; Eisen, J.A. PhyloSift: phylogenetic analysis of genomes and metagenomes. PeerJ 2014, 2, e243. [Google Scholar]
- Hyatt, D.; Chen, G.-L.; LoCascio, P.F.; Land, M.L.; Larimer, F.W.; Hauser, L.J. Prodigal: Prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 2010, 11, 119. [Google Scholar] [CrossRef]
- Moriya, Y.; Itoh, M.; Okuda, S.; Yoshizawa, A.C.; Kanehisa, M. KAAS: An automatic genome annotation and pathway reconstruction server. Nucleic Acids Res. 2007, 35, W182–W185. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef]
- Capella-Gutiérrez, S.; Silla-Martínez, J.M.; Gabaldón, T. trimAl: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive tree of life (iTOL) v3: An online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res. 2016, 44, W242–W245. [Google Scholar] [CrossRef]
- Lowe, T.M.; Chan, P.P. tRNAscan-SE On-line: Integrating search and context for analysis of transfer RNA genes. Nucleic Acids Res. 2016, 44, W54–W57. [Google Scholar]
- Grant, J.R.; Stothard, P. The CGView Server: A comparative genomics tool for circular genomes. Nucleic Acids Res. 2008, 36, W181–W184. [Google Scholar] [CrossRef]
- Cattolico, R.A.; Jacobs, M.A.; Zhou, Y.; Chang, J.; Duplessis, M.; Lybrand, T.; McKay, J.; Ong, H.C.; Sims, E.; Rocap, G. Chloroplast genome sequencing analysis of Heterosigma akashiwo CCMP452 (West Atlantic) and NIES293 (West Pacific) strains. Bmc Genomics 2008, 9, 211. [Google Scholar] [CrossRef]
- Seoane, S.; Hyodo, K.; Ueki, S. Chloroplast Genome Sequences of Seven Strains of the Bloom-Forming Raphidophyte Heterosigma akashiwo. Genome Announc. 2017, 5, e01030-17. [Google Scholar] [CrossRef]
- Hunsperger, H.M.; Randhawa, T.; Cattolico, R.A. Extensive horizontal gene transfer, duplication, and loss of chlorophyll synthesis genes in the algae. BMC Evol. Biol. 2015, 15, 16. [Google Scholar]
- Sassenhagen, I.; Sefbom, J.; Sall, T.; Godhe, A.; Rengefors, K. Freshwater protists do not go with the flow: Population structure in Gonyostomum semen independent of connectivity among lakes. Environ. Microbiol. 2015, 17, 5063–5072. [Google Scholar]
- de Bourcy, C.F.A.; Vlaminck, I.D.; Kanbar, J.N.; Wang, J.; Gawad, C.; Quake, S.R. A Quantitative Comparison of Single-Cell Whole Genome Amplification Methods. PLOS ONE 2014, 9, e105585. [Google Scholar]
- Borgström, E.; Paterlini, M.; Mold, J.E.; Frisen, J.; Lundeberg, J. Comparison of whole genome amplification techniques for human single cell exome sequencing. PLOS ONE 2017, 12, e0171566. [Google Scholar]
- Marcy, Y.; Ishoey, T.; Lasken, R.S.; Stockwell, T.B.; Walenz, B.P.; Halpern, A.L.; Beeson, K.Y.; Goldberg, S.M.D.; Quake, S.R. Nanoliter Reactors Improve Multiple Displacement Amplification of Genomes from Single Cells. PLOS Genet. 2007, 3, e155. [Google Scholar]
- Fu, Y.; Li, C.; Lu, S.; Zhou, W.; Tang, F.; Xie, X.S.; Huang, Y. Uniform and accurate single-cell sequencing based on emulsion whole-genome amplification. Proc. Natl. Acad. Sci. USA 2015, 112, 11923–11928. [Google Scholar]
- Zong, C.; Lu, S.; Chapman, A.R.; Xie, X.S. Genome-Wide Detection of Single-Nucleotide and Copy-Number Variations of a Single Human Cell. Science 2012, 338, 1622–1626. [Google Scholar] [CrossRef]
- Stepanauskas, R.; Fergusson, E.A.; Brown, J.; Poulton, N.J.; Tupper, B.; Labonté, J.M.; Becraft, E.D.; Brown, J.M.; Pachiadaki, M.G.; Povilaitis, T.; et al. Improved genome recovery and integrated cell-size analyses of individual uncultured microbial cells and viral particles. Nat. Commun. 2017, 8, 84. [Google Scholar] [CrossRef]
- Derelle, R.; López-García, P.; Timpano, H.; Moreira, D. A Phylogenomic Framework to Study the Diversity and Evolution of Stramenopiles (=Heterokonts). Mol. Biol. Evol. 2016, 33, 2890–2898. [Google Scholar]
- Li, H.; Wang, W.; Wang, Z.; Lin, X.; Zhang, F.; Yang, L. De novo transcriptome analysis of carotenoid and polyunsaturated fatty acid metabolism in Rhodomonas sp. J. Appl. Phycol. 2016, 28, 1649–1656. [Google Scholar]
- Takaichi, S. Carotenoids in algae: Distributions, biosyntheses and functions. Mar. Drugs 2011, 9, 1101–1118. [Google Scholar]
- Takaichi, S.; Yokoyama, A.; Mochimaru, M.; Uchida, H.; Murakami, A. Carotenogenesis diversification in phylogenetic lineages of Rhodophyta. J. Phycol. 2016, 52, 329–338. [Google Scholar]
- Swift, I.E.; Milborrow, B.V.; Jeffrey, S.W. Formation of Neoxanthin, Diadinoxanthin and Peridinin from [14C]Zeaxanthin by a cell-free System from Ampidinium carterae. Phytochemistry 1982, 21, 2859–2864. [Google Scholar] [CrossRef]
- Race, H.L.; Herrmann, R.G.; Martin, W. Why have organelles retained genomes? Trends Genet. 1999, 15, 364–370. [Google Scholar] [CrossRef]
- Leister, D. Chloroplast research in the genomic age. Trends Genet. 2003, 19, 47–56. [Google Scholar] [CrossRef]
- Soll, J.; Schleiff, E. Protein import into chloroplasts. Nat. Rev. Mol. Cell Biol. 2004, 5, 198–208. [Google Scholar] [CrossRef] [PubMed]
- Fong, A.; Archibald, J.M. Evolutionary Dynamics of Light-Independent Protochlorophyllide Oxidoreductase Genes in the Secondary Plastids of Cryptophyte Algae. Eukaryot. Cell 2008, 7, 550–553. [Google Scholar] [CrossRef] [PubMed]
- Björnerås, C.; Weyhenmeyer, G.A.; Evans, C.D.; Gessner, M.O.; Grossart, H.-P.; Kangur, K.; Kokorite, I.; Kortelainen, P.; Laudon, H.; Lehtoranta, J.; et al. Widespread Increases in Iron Concentration in European and North American Freshwaters. Glob. Biogeochem. Cycles 2017, 31, 1488–1500. [Google Scholar] [CrossRef]
- Yamazaki, S.; Nomata, J.; Fujita, Y. Differential Operation of Dual Protochlorophyllide Reductases for Chlorophyll Biosynthesis in Response to Environmental Oxygen Levels in the Cyanobacterium Leptolyngbya boryana. Plant Physiol. 2006, 142, 911–922. [Google Scholar] [PubMed]
- Salonen, K.; Rosenberg, M. Advantages from diel vertical migration can explain the dominance of Gonyostomum semen (Raphidophyceae) in a small, steeply-stratified humic lake. J. Plankton Res. 2000, 22, 1841–1853. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| G. semen | V. virescens | |
|---|---|---|
| #reads (fwd and rev) | 1,409,520 | 790,172 |
| % bacterial reads | 81.35 | 85.05 |
| mean coverage of all contigs | 95.3 | 75.4 |
| #chl contigs | 39 | 35 |
| chl contigs length (bp) | 74,603 | 88,279 |
| mean coverage of chl contigs | 612.9 | 472.3 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sassenhagen, I.; Rengefors, K. Sequencing and Phylogenetic Analysis of Chloroplast Genes in Freshwater Raphidophytes. Genes 2019, 10, 245. https://doi.org/10.3390/genes10030245
Sassenhagen I, Rengefors K. Sequencing and Phylogenetic Analysis of Chloroplast Genes in Freshwater Raphidophytes. Genes. 2019; 10(3):245. https://doi.org/10.3390/genes10030245
Chicago/Turabian StyleSassenhagen, Ingrid, and Karin Rengefors. 2019. "Sequencing and Phylogenetic Analysis of Chloroplast Genes in Freshwater Raphidophytes" Genes 10, no. 3: 245. https://doi.org/10.3390/genes10030245