Group Lasso Regularized Deep Learning for Cancer Prognosis from Multi-Omics and Clinical Features

 ,
,
Abstract
:1. Introduction
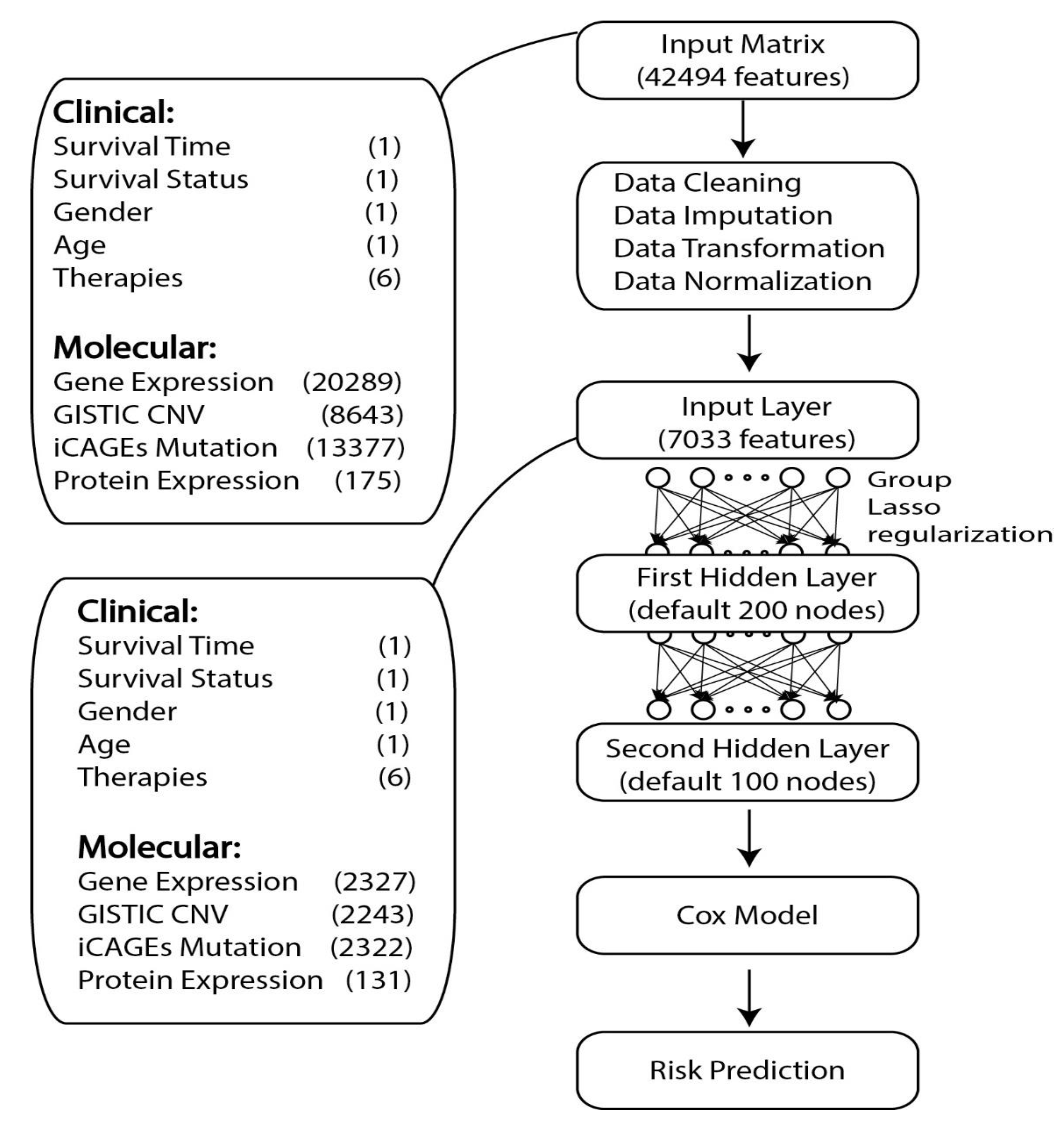
2. Materials and Methods
2.1. Data Collection
2.2. TCGA Data Preprocessing
2.3. Data Simulation
2.4. GDP Model
2.5. Model Training
2.6. Model Evaluation and Feature Selection
2.7. Availabilities of Software
3. Results
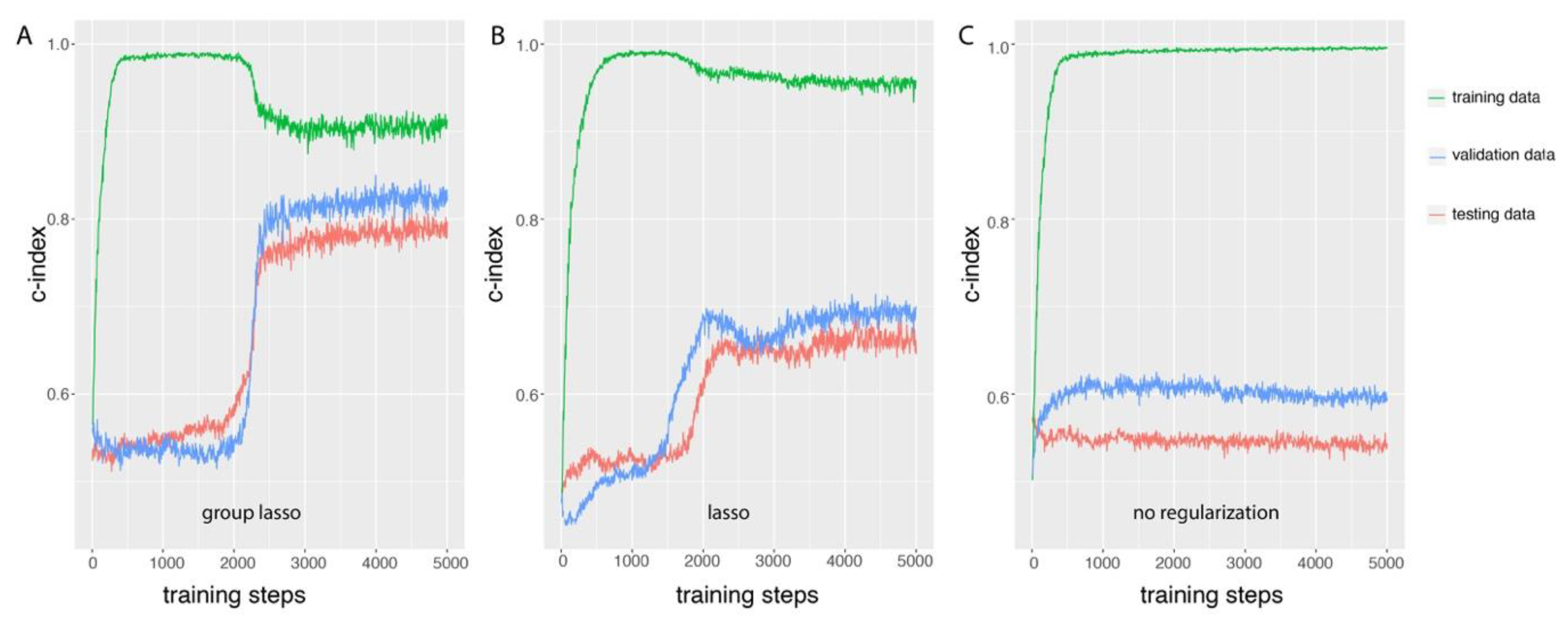
3.1. Group Lasso Prevents Overfitting During GDP Training
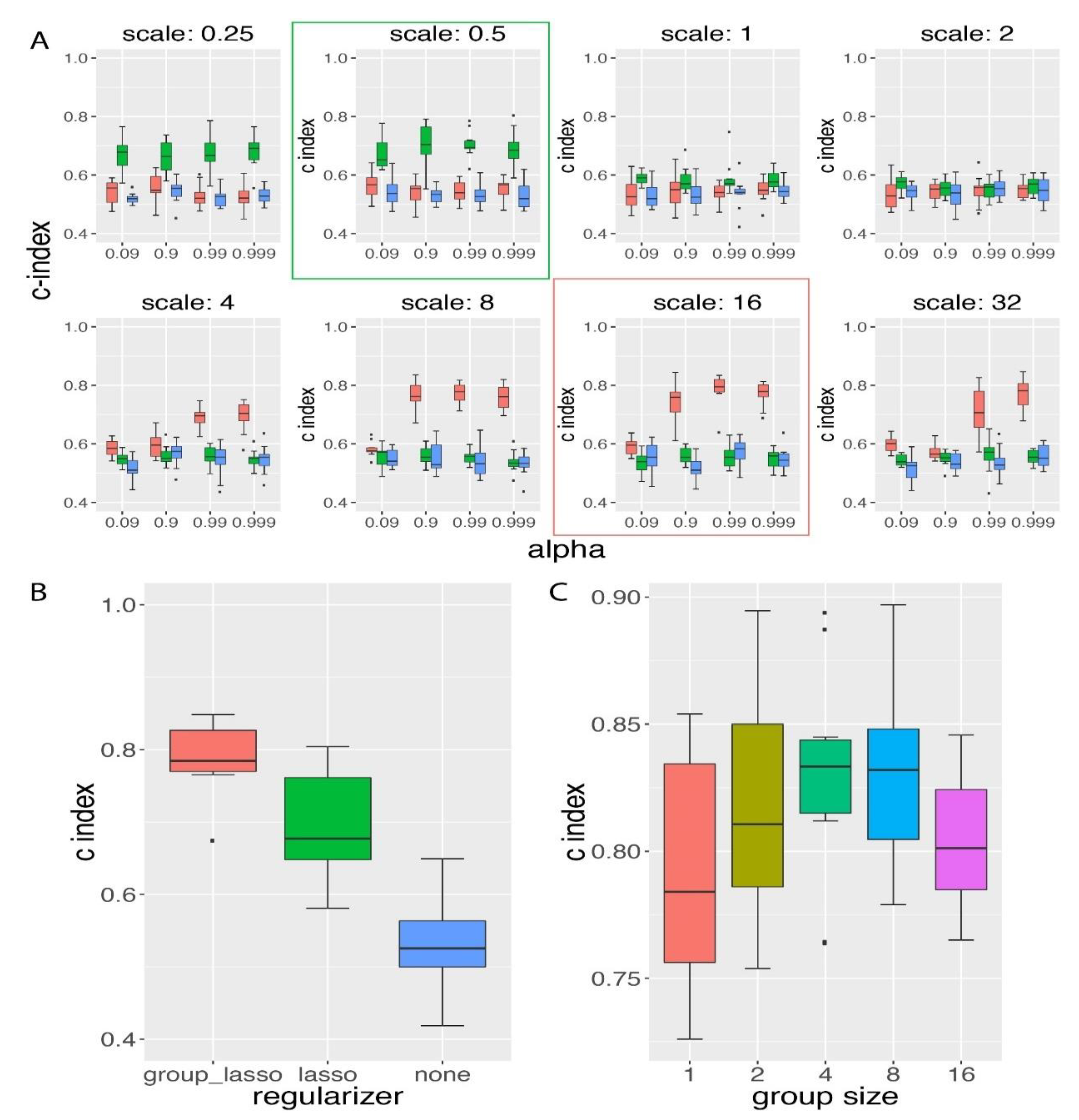
3.2. Group Lasso Performed Better than Lasso Regularization on the Simulated Time-To-Event Data with Group Prior Knowledge
3.3. Influence of Group Size on the Performance of GDP Survival Prediction
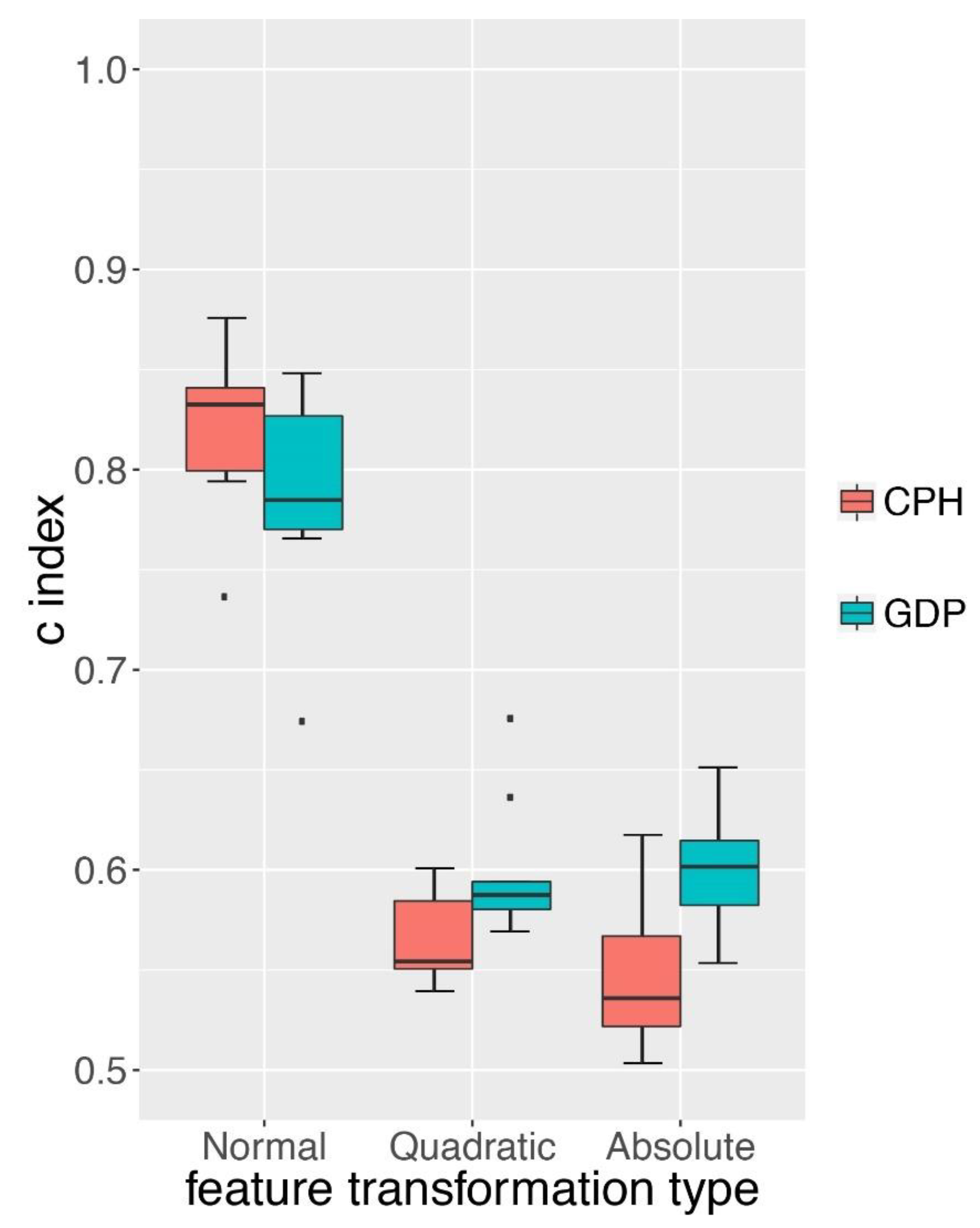
3.4. GDP Performed Better than CPH under Complex Simulations
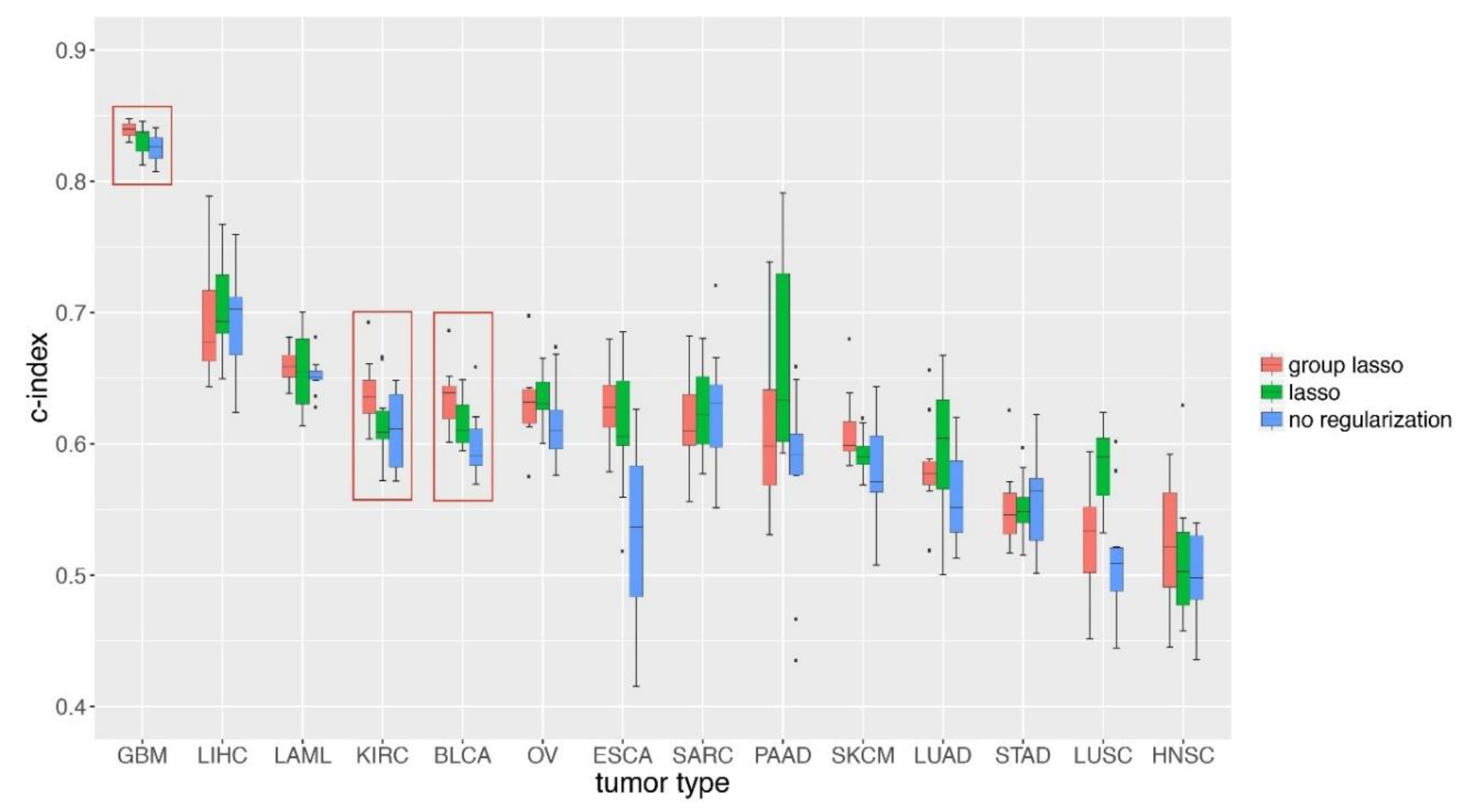
3.5. GDP Performances on TCGA Cancer Data
4. Discussions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| Lasso | Least absolute shrinkage and selection operator |
| TCGA | The Cancer Genome Atlas |
| C-index | Concordance index |
| CPH | Cox proportional hazard model |
| NSS | Non-linear survival simulation |
| LSS | Linear survival simulation |
| CNV | Copy number variation |
| GDP | Group lasso regularized deep learning for the survival prediction in cancer patients |
| TRIPOD | Transparent Reporting of a multivariable prediction model for Individual Prognosis Or Diagnosis |
| iCAGES | Integrated CAncer Genome Score |
| GISTIC | Genomic Identification of Significant Targets in Cancer |
| GDAC | Genome Data Analysis Center |
References
- James, N.D.; Sydes, M.R.; Clarke, N.W.; Mason, M.D.; Dearnaley, D.P.; Spears, M.R.; Ritchie, A.W.S.; Parker, C.C.; Russell, J.M.; Attard, G.; et al. Addition of docetaxel, zoledronic acid, or both to first-line long-term hormone therapy in prostate cancer (STAMPEDE): Survival results from an adaptive, multiarm, multistage, platform randomised controlled trial. Lancet 2016, 387, 1163–1177. [Google Scholar] [CrossRef]
- Von Minckwitz, G.; Procter, M.; de Azambuja, E.; Zardavas, D.; Benyunes, M.; Viale, G.; Suter, T.; Arahmani, A.; Rouchet, N.; Clark, E.; et al. Adjuvant pertuzumab and trastuzumab in early HER2-positive breast cancer. N. Engl. J. Med. 2017, 377, 122–131. [Google Scholar] [CrossRef] [PubMed]
- Mlecnik, B.; Bindea, G.; Angell, H.K.; Maby, P.; Angelova, M.; Tougeron, D.; Church, S.E.; Lafontaine, L.; Fischer, M.; Fredriksen, T.; et al. Integrative analyses of colorectal cancer show immunoscore is a stronger predictor of patient survival than microsatellite instability. Immunity 2016, 44, 698–711. [Google Scholar] [CrossRef] [PubMed]
- Flynn, R. Survival analysis. J. Clin. Nurs. 2012, 21, 2789–2797. [Google Scholar] [CrossRef]
- Cox, D.R. Regression models and life-tables. J. R. Stat. Soc. B 1972, 34, 187. [Google Scholar] [CrossRef]
- Goodwin, S.; McPherson, J.D.; McCombie, W.R. Coming of age: Ten years of next-generation sequencing technologies. Nat. Rev. Genet. 2016, 17, 333–351. [Google Scholar] [CrossRef]
- Weinstein, J.N.; Collisson, E.A.; Mills, G.B.; Shaw, K.R.M.; Ozenberger, B.A.; Ellrott, K.; Shmulevich, I.; Sander, C.; Stuart, J.M. Network CGAR: The cancer genome atlas pan-cancer analysis project. Nature Genet. 2013, 45, 1113–1120. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research Network. Electronic address edsc, cancer genome atlas research N: Comprehensive and integrated genomic characterization of adult soft tissue sarcomas. Cell 2017, 171, 950–965. [Google Scholar] [CrossRef]
- Tibshirani, R.; Witten, D.M. Survival analysis with high-dimensional covariates. Stat. Methods Med. R. 2010, 19, 29–51. [Google Scholar]
- Tibshirani, R. Regression shrinkage and selection via the lasso. J. R. Stat. Soc. 1996, 58, 267–288. [Google Scholar] [CrossRef]
- Tibshirani, R. The lasso method for variable selection in the Cox model. Stat. Med. 1997, 16, 385–395. [Google Scholar] [CrossRef]
- Zou, H.; Hastie, T. Regularization and variable selection via the elastic net. J. R. Stat. Soc. 2005, 67, 301–320. [Google Scholar] [CrossRef]
- Werner, H.M.; Mills, G.B.; Ram, P.T. Cancer systems biology: A peek into the future of patient care? Nat. Rev. Clin. Oncol. 2014, 11, 167–176. [Google Scholar] [CrossRef]
- Yuan, M.; Lin, Y. Model selection and estimation in regression with grouped variables. J. R. Stat. Soc B 2006, 68, 49–67. [Google Scholar] [CrossRef] [Green Version]
- Meier, L.; van de Geer, S.A.; Buhlmann, P. The group lasso for logistic regression. J. R. Stat. Soc. B 2008, 70, 53–71. [Google Scholar] [CrossRef] [Green Version]
- LeCun, Y.; Bengio, Y.; Hinton, G. Deep learning. Nature 2015, 521, 436–444. [Google Scholar] [CrossRef] [PubMed]
- Silver, D.; Huang, A.; Maddison, C.J.; Guez, A.; Sifre, L.; van den Driessche, G.; Schrittwieser, J.; Antonoglou, I.; Panneershelvam, V.; Lanctot, M.; et al. Mastering the game of go with deep neural networks and tree search. Nature 2016, 529, 484–489. [Google Scholar] [CrossRef]
- Karpathy, A.; Fei-Fei, L. Deep visual-semantic alignments for generating image descriptions. IEEE Trans. Pattern Anal. Mach. Intell. 2017, 39, 664–676. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Seff, A.; Kornhauser, A.; Xiao, J. DeepDriving: Learning affordance for direct perception in autonomous driving. In Proceedings of the 2015 IEEE International Conference on Computer Vision (ICCV), Las Condes, Chile, 7–13 December 2015. [Google Scholar]
- Alipanahi, B.; Delong, A.; Weirauch, M.T.; Frey, B.J. Predicting the sequence specificities of DNA- and RNA-binding proteins by deep learning. Nat. Biotechnol. 2015, 33, 831. [Google Scholar] [CrossRef] [PubMed]
- Xiong, H.Y.; Alipanahi, B.; Lee, L.J.; Bretschneider, H.; Merico, D.; Yuen, R.K.C.; Hua, Y.M.; Gueroussov, S.; Najafabadi, H.S.; Hughes, T.R.; et al. The human splicing code reveals new insights into the genetic determinants of disease. Science 2015, 347, 6218. [Google Scholar] [CrossRef]
- Zhou, J.; Troyanskaya, O.G. Predicting effects of noncoding variants with deep learning-based sequence model. Nat. Methods 2015, 12, 931–934. [Google Scholar] [CrossRef]
- Katzman, J.L.; Shaham, U.; Cloninger, A.; Bates, J.; Jiang, T.; Kluger, Y. DeepSurv: Personalized Treatment Recommender System Using A Cox Proportional Hazards Deep Neural Network. BMC Med. Res. Methodol. 2016, 18, 24. [Google Scholar] [CrossRef] [PubMed]
- Yousefi, S.; Amrollahi, F.; Amgad, M.; Dong, C.; Lewis, J.E.; Song, C.; Gutman, D.A.; Halani, S.H.; Vega, J.E.V.; Brat, D.J.; et al. Predicting clinical outcomes from large scale cancer genomic profiles with deep survival models. Sci. Rep. 2017, 7, 11707. [Google Scholar] [CrossRef]
- Martín Abadi, A.A.; Paul, B.; Brevdo, E.; Zhifeng, C.; Craig, C.; Greg, S.; Corrado, A.D.; Jeffrey, D.; Devin, M.; Sanjay, G. Google research: TensorFlow: Large-scale machine learning on heterogeneous distributed systems. arXiv, 2016; arXiv:1603.04467. [Google Scholar]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [PubMed]
- Mermel, C.H.; Schumacher, S.E.; Hill, B.; Meyerson, M.L.; Beroukhim, R.; Getz, G. GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol. 2011, 12, R41. [Google Scholar] [CrossRef]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef]
- Dong, C.; Guo, Y.; Yang, H.; He, Z.; Liu, X.; Wang, K. ICAGES: Integrated cancer genome score for comprehensively prioritizing driver genes in personal cancer genomes. Genome Med. 2016, 8, 135. [Google Scholar] [CrossRef]
- Bender, R.; Augustin, T.; Blettner, M. Generating survival times to simulate COX proportional hazards models. Stat. Med. 2005, 24, 1713–1723. [Google Scholar] [CrossRef]
- Harrell, F.E., Jr.; Califf, R.M.; Pryor, D.B.; Lee, K.L.; Rosati, R.A. Evaluating the yield of medical tests. JAMA 1982, 247, 2543–2546. [Google Scholar] [CrossRef]
- Helbing, C.C.; Veillette, C.; Riabowol, K.; Johnston, R.N.; Garkavtsev, I. A novel candidate tumor suppressor, ING1, is involved in the regulation of apoptosis. Cancer Res. 1997, 57, 1255–1258. [Google Scholar]
- Tallen, U.G.; Truss, M.; Kunitz, F.; Wellmann, S.; Unryn, B.; Sinn, B.; Lass, U.; Krabbe, S.; Holtkamp, N.; Hagemeier, C.; et al. Down-regulation of the inhibitor of growth 1 (ING1) tumor suppressor sensitizes p53-deficient glioblastoma cells to cisplatin-induced cell death. J. Neurooncol. 2008, 86, 23–30. [Google Scholar] [CrossRef]
- Yuan, Y.; van Allen, E.M.; Omberg, L.; Wagle, N.; Amin-Mansour, A.; Sokolov, A.; Byers, L.A.; Xu, Y.; Hess, K.R.; Diao, L.; et al. Assessing the clinical utility of cancer genomic and proteomic data across tumor types. Nat. Biotechnol. 2014, 32, 644–652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simon, N.; Friedman, J.; Hastie, T.; Tibshirani, R. A sparse-group lasso. J. Comput. Graph. Stat. 2013, 22, 231–245. [Google Scholar] [CrossRef]
- Collins, G.S.; Reitsma, J.B.; Altman, D.G.; Moons, K.G. Transparent reporting of a multivariable prediction model for individual prognosis or diagnosis (TRIPOD). Ann. Intern. Med. 2015, 162, 735–736. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: The R scripts for survival data simulation and the GDP python package can be found in GitHub (https://github.com/WGLab/GDP). |






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tumor Name | Tumor Full Name | # of Patients | # Censored |
|---|---|---|---|
| GBM | Glioblastoma multiforme | 579 | 101 |
| OV | Ovarian serous cystadenocarcinoma | 571 | 232 |
| KIRC | Kidney renal clear cell carcinoma | 532 | 355 |
| HNSC | Head and neck squamous cell carcinoma | 528 | 304 |
| LUAD | Lung adenocarcinoma | 507 | 322 |
| LUSC | Lung squamous cell carcinoma | 504 | 284 |
| SKCM | Skin cutaneous melanoma | 469 | 249 |
| STAD | Stomach adenocarcinoma | 443 | 270 |
| BLCA | Bladder urothelial carcinoma | 409 | 229 |
| LIHC | Liver hepatocellular carcinoma | 377 | 245 |
| SARC | Sarcoma | 261 | 162 |
| LAML | Acute myeloid leukemia | 198 | 66 |
| PAAD | Pancreatic adenocarcinoma | 185 | 85 |
| ESCA | Esophageal carcinoma | 185 | 108 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xie, G.; Dong, C.; Kong, Y.; Zhong, J.F.; Li, M.; Wang, K. Group Lasso Regularized Deep Learning for Cancer Prognosis from Multi-Omics and Clinical Features. Genes 2019, 10, 240. https://doi.org/10.3390/genes10030240
Xie G, Dong C, Kong Y, Zhong JF, Li M, Wang K. Group Lasso Regularized Deep Learning for Cancer Prognosis from Multi-Omics and Clinical Features. Genes. 2019; 10(3):240. https://doi.org/10.3390/genes10030240
Chicago/Turabian StyleXie, Gangcai, Chengliang Dong, Yinfei Kong, Jiang F. Zhong, Mingyao Li, and Kai Wang. 2019. "Group Lasso Regularized Deep Learning for Cancer Prognosis from Multi-Omics and Clinical Features" Genes 10, no. 3: 240. https://doi.org/10.3390/genes10030240