Exceptional Longevity and Polygenic Risk for Cardiovascular Health


 , , , , , and
, , , , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Participants
2.2. Genotyping
2.3. Polygenic Risk Scores (PRS)
2.4. Statistical Analyses
3. Results
3.1. Sample Characteristics
3.2. Polygenic Risk Scores (PRS)
3.3. PRS Associations with Measured Phenotypes
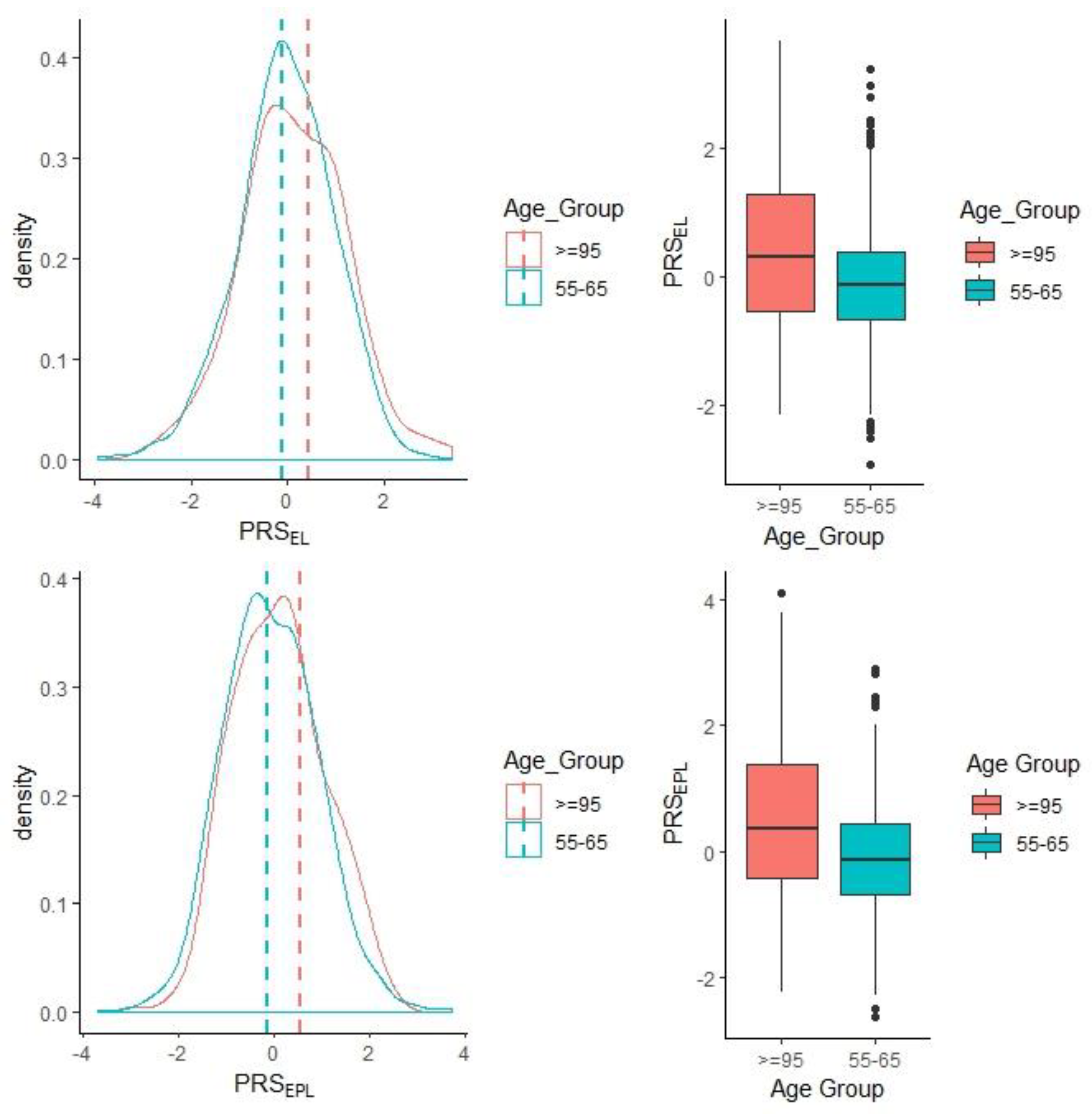
3.3.1. Longevity PRS with Exceptional Longevity
3.3.2. Association of Cardiovascular Risk Factor (Lipids) PRS with Measured Lipid Levels
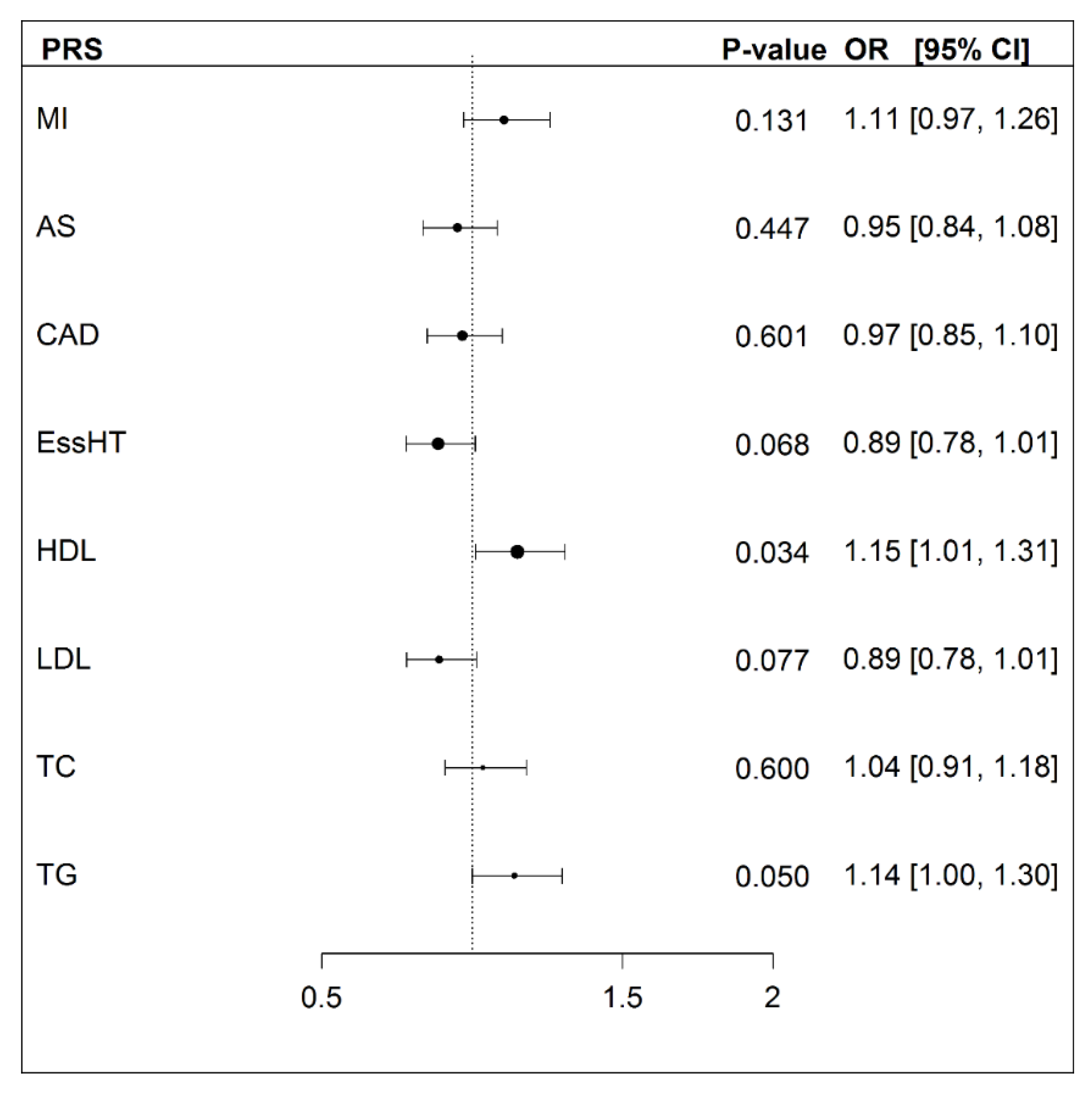
3.3.3. Cardiovascular Health PRS and Exceptional Longevity
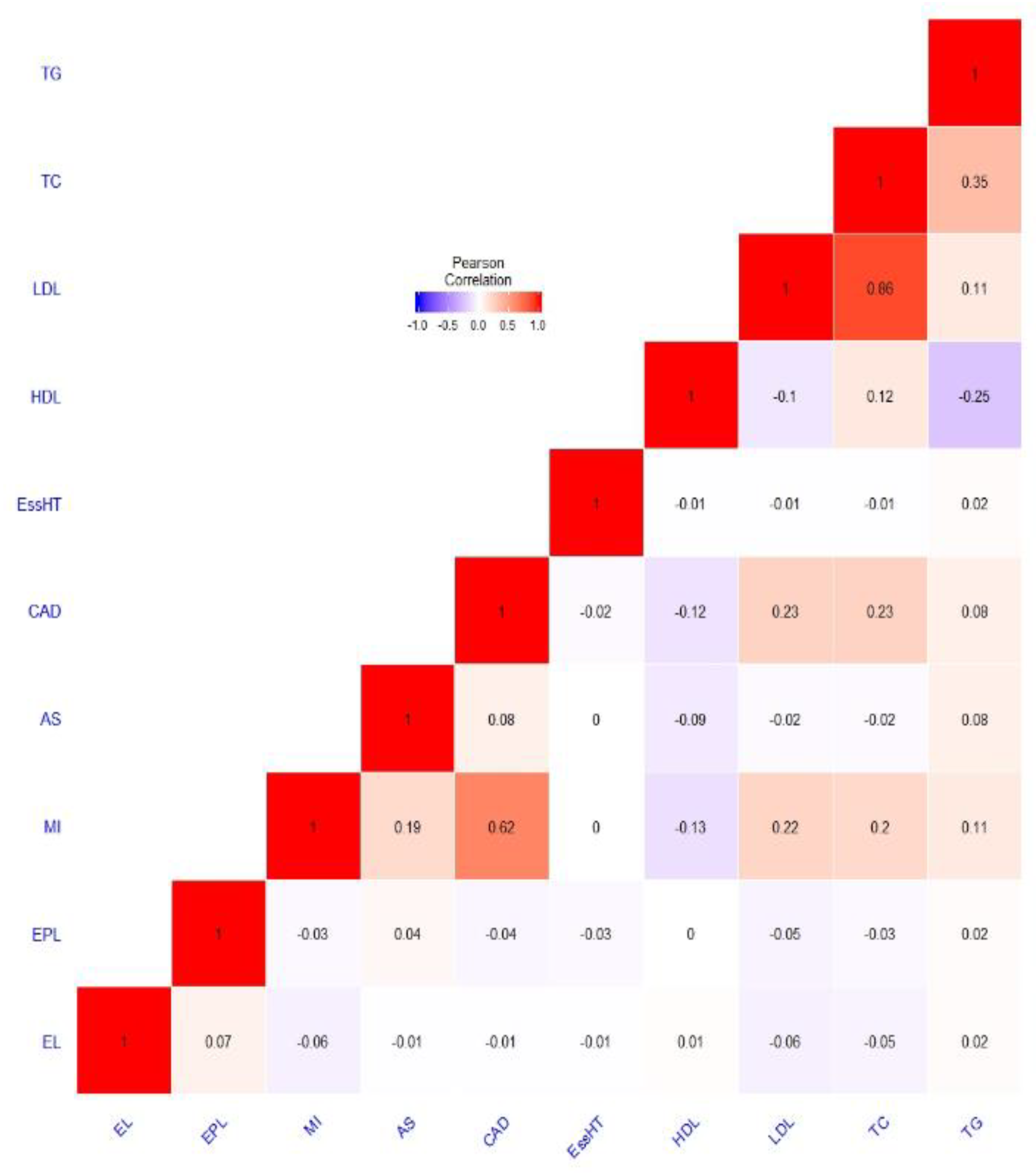
PRS Genetic Overlap
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Oeppen, J.; Vaupel, J.W. Demography. Broken limits to life expectancy. Science 2002, 296, 1029–1031. [Google Scholar] [CrossRef]
- Fries, J.F. Aging, natural death, and the compression of morbidity. 1980. Bull. World Health Organ 2002, 80, 245–250. [Google Scholar]
- Franceschi, C.; Passarino, G.; Mari, D.; Monti, D. Centenarians as a 21st century healthy aging model: A legacy of humanity and the need for a world-wide consortium (WWC100+). Mech. Ageing Dev. 2017, 165, 55–58. [Google Scholar] [CrossRef]
- Atkins, J.L.; Delgado, J.; Pilling, L.C.; Bowman, K.; Masoli, J.A.H.; Kuchel, G.A.; Ferrucci, L.; Melzer, D. Impact of Low Cardiovascular Risk Profiles on Geriatric Outcomes: Evidence From 421,000 Participants in Two Cohorts. J. Gerontol. A Biol. Sci. Med. Sci. 2018. [Google Scholar] [CrossRef]
- Vaarhorst, A.A.; Beekman, M.; Suchiman, E.H.; van Heemst, D.; Houwing-Duistermaat, J.J.; Westendorp, R.G.; Slagboom, P.E.; Heijmans, B.T. Leiden Longevity Study Group. Lipid metabolism in long-lived families: The Leiden Longevity Study. Age 2011, 33, 219–227. [Google Scholar] [CrossRef] [PubMed]
- Barzilai, N.; Atzmon, G.; Schechter, C.; Schaefer, E.J.; Cupples, A.L.; Lipton, R.; Cheng, S.; Shuldiner, A.R. Unique lipoprotein phenotype and genotype associated with exceptional longevity. JAMA 2003, 290, 2030–2040. [Google Scholar] [CrossRef] [PubMed]
- Muenchhoff, J.; Song, F.; Poljak, A.; Crawford, J.D.; Mather, K.A.; Kochan, N.A.; Yang, Z.; Trollor, J.N.; Reppermund, S.; Maston, K.; et al. Plasma apolipoproteins and physical and cognitive health in very old individuals. Neurobiol. Aging 2017, 55, 49–60. [Google Scholar] [CrossRef]
- Pilling, L.C.; Atkins, J.L.; Bowman, K.; Jones, S.E.; Tyrrell, J.; Beaumont, R.N.; Ruth, K.S.; Tuke, H.; Yaghootkar, M.A.; Wood, A.R.; et al. Human longevity is influenced by many genetic variants: Evidence from 75,000 UK Biobank participants. Aging 2016, 8, 547–560. [Google Scholar] [CrossRef] [PubMed]
- Revelas, M.; Thalamuthu, A.; Oldmeadow, C.; Evans, T.J.; Armstrong, N.J.; Kwok, J.B.; Brodaty, H.; Schofield, P.R.; Scott, R.J.; Sachdev, P.S.; et al. Review and meta-analysis of genetic polymorphisms associated with exceptional human longevity. Mech. Ageing Dev. 2018, 175, 24–34. [Google Scholar] [CrossRef]
- Dominiczak, M.H.; Caslake, M.J. Apolipoproteins: Metabolic role and clinical biochemistry applications. Ann. Clin. Biochem. 2011, 48, 498–515. [Google Scholar] [CrossRef]
- Lahoz, C.; Schaefer, E.J.; Cupples, L.A.; Wilson, P.W.; Levy, D.; Osgood, D.; Parpos, S.; Pedro-Botet, J.; Daly, J.A.; Ordovas, J.M. Apolipoprotein E genotype and cardiovascular disease in the Framingham Heart Study. Atherosclerosis 2001, 154, 529–537. [Google Scholar] [CrossRef]
- Morris, B.J.; Willcox, D.C.; Donlon, T.A.; Willcox, B.J. FOXO3: A Major Gene for Human Longevity—A Mini-Review. Gerontology 2015, 61, 515–525. [Google Scholar] [CrossRef] [PubMed]
- Willcox, B.J.; Morris, B.J.; Tranah, G.J.; Chen, R.; Masaki, K.H.; He, Q.; Willcox, D.C.; Allsopp, R.C.; Moisyadi, S.; Gerschenson, M.; et al. Longevity-Associated FOXO3 Genotype and its Impact on Coronary Artery Disease Mortality in Japanese, Whites, and Blacks: A Prospective Study of Three American Populations. J. Gerontol. A Biol. Sci. Med. Sci. 2017, 72, 724–728. [Google Scholar] [CrossRef] [PubMed]
- Vergani, C.; Lucchi, T.; Caloni, M.; Ceconi, I.; Calabresi, C.; Scurati, S.; Arosio, B. I405V polymorphism of the cholesteryl ester transfer protein (CETP) gene in young and very old people. Arch. Gerontol. Geriat. 2006, 43, 213–221. [Google Scholar] [CrossRef] [PubMed]
- Atzmon, G.; Rincon, M.; Schechter, C.B.; Shuldiner, A.R.; Lipton, R.B.; Bergman, A.; Barzilai, N. Lipoprotein genotype and conserved pathway for exceptional longevity in humans. PLoS Biol. 2006, 4, e113. [Google Scholar] [CrossRef]
- Panza, F.; D’Introno, A.; Colacicco, A.M.; Capurso, C.; Capurso, S.; Kehoe, P.G.; Capurso, A.; Solfrizzi, V. Vascular genetic factors and human longevity. Mech. Ageing Dev. 2004, 125, 169–178. [Google Scholar] [CrossRef]
- Sachdev, P.S.; Levitan, C.; Crawford, J.; Sidhu, M.; Slavin, M.; Richmond, R.; Kochan, N.; Brodaty, H.; Wen, W.; Kang, K.; et al. The Sydney Centenarian Study: Methodology and profile of centenarians and near-centenarians. Int. Psychogeriatr. 2013, 25, 993–1005. [Google Scholar] [CrossRef] [PubMed]
- Sachdev, P.S.; Brodaty, H.; Reppermund, S.; Kochan, N.A.; Trollor, J.N.; Draper, B.; Slavin, M.J.; Crawford, J.; Kang, K.; Broe, G.A.; et al. The Sydney Memory and Ageing Study (MAS): Methodology and baseline medical and neuropsychiatric characteristics of an elderly epidemiological non-demented cohort of Australians aged 70-90 years. Int. Psychogeriatr. 2010, 22, 1248–1264. [Google Scholar] [CrossRef]
- McEvoy, M.; Smith, W.; D’Este, C.; Duke, J.; Peel, R.; Schofield, P.; Scott, R.; Byles, J.; Henry, D.; Ewald, B.; et al. Cohort profile: The Hunter Community Study. Int. J. Epidemiol. 2010, 39, 1452–1463. [Google Scholar] [CrossRef]
- Song, F.; Poljak, A.; Crawford, J.; Kochan, N.A.; Wen, W.; Cameron, B.; Lux, O.; Brodaty, H.; Mather, K.; Smythe, G.A.; et al. Plasma apolipoprotein levels are associated with cognitive status and decline in a community cohort of older individuals. PLoS ONE 2012, 7, e34078. [Google Scholar] [CrossRef] [PubMed]
- Muenchhoff, J.; Poljak, A.; Song, F.; Raftery, M.; Brodaty, H.; Duncan, M.; McEvoy, M.; Attia, J.; Schofield, P.W.; Sachdev, P.S. Plasma protein profiling of mild cognitive impairment and Alzheimer’s disease across two independent cohorts. J. Alzheimers Dis. 2015, 43, 1355–1373. [Google Scholar] [CrossRef]
- Price, A.L.; Patterson, N.J.; Plenge, R.M.; Weinblatt, M.E.; Shadick, N.A.; Reich, D. Principal components analysis corrects for stratification in genome-wide association studies. Nat. Genet. 2006, 38, 904–909. [Google Scholar] [CrossRef]
- Das, S.; Forer, L.; Schonherr, S.; Sidore, C.; Locke, A.E.; Kwong, A.; Vrieze, S.I.; Chew, E.Y.; Levy, S.; McGue, M.; et al. Next-generation genotype imputation service and methods. Nat. Genet. 2016, 48, 1284–1287. [Google Scholar] [CrossRef]
- Oldmeadow, C.; Holliday, E.G.; McEvoy, M.; Scott, R.; Kwok, J.B.; Mather, K.; Sachdev, P.; Schofield, P.; Attia, J. Concordance between direct and imputed APOE genotypes using 1000 Genomes data. J. Alzheimers Dis. 2014, 42, 391–393. [Google Scholar] [CrossRef] [PubMed]
- Euesden, J.; Lewis, C.M.; O’Reilly, P.F. PRSice: Polygenic Risk Score software. Bioinformatics 2015, 31, 1466–1468. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. R: A Language and Environment for Statistical Computing; R Core Team: Vienna, Austria, 2017. [Google Scholar]
- Nagelkerke, N.J.D. A Note on a General Definition of the Coefficient of Determination. Biometrika 1991, 78, 691–692. [Google Scholar] [CrossRef]
- Zhang, D. rsq: R-Squared and Related Measures, R Package version 1.1; Vienna, Austria, 2018. [Google Scholar]
- Zhang, M. WebPower: Basic and Advanced Statistical Power Analysis, R Package version 0.5.2; Vienna, Austria, 2018. [Google Scholar]
- Broer, L.; Buchman, A.S.; Deelen, J.; Evans, D.S.; Faul, J.D.; Lunetta, K.L.; Sebastiani, P.; Smith, J.A.; Smith, A.V.; Tanaka, T.; et al. GWAS of longevity in CHARGE consortium confirms APOE and FOXO3 candidacy. J. Gerontol. A Biol. Sci. Med. Sci. 2015, 70, 110–118. [Google Scholar] [CrossRef]
- Myocardial Infarction, G.; Investigators, C.A.E.C.; Stitziel, N.O.; Stirrups, K.E.; Masca, N.G.; Erdmann, J.; Ferrario, P.G.; Konig, I.R.; Weeke, P.E.; Webb, T.R.; et al. Coding Variation in ANGPTL4, LPL, and SVEP1 and the Risk of Coronary Disease. N. Engl. J. Med. 2016, 374, 1134–1144. [Google Scholar] [CrossRef]
- Malik, R.; Chauhan, G.; Traylor, M.; Sargurupremraj, M.; Okada, Y.; Mishra, A.; Rutten-Jacobs, L.; Giese, A.K.; van der Laan, S.W.; Gretarsdottir, S.; et al. Multiancestry genome-wide association study of 520,000 subjects identifies 32 loci associated with stroke and stroke subtypes. Nat. Genet. 2018, 50, 524–537. [Google Scholar] [CrossRef] [PubMed]
- Nelson, C.P.; Goel, A.; Butterworth, A.S.; Kanoni, S.; Webb, T.R.; Marouli, E.; Zeng, L.; Ntalla, I.; Lai, F.Y.; Hopewell, J.C.; et al. Association analyses based on false discovery rate implicate new loci for coronary artery disease. Nat. Genet. 2017, 49, 1385–1391. [Google Scholar] [CrossRef] [PubMed]
- Willer, C.J.; Schmidt, E.M.; Sengupta, S.; Peloso, G.M.; Gustafsson, S.; Kanoni, S.; Ganna, A.; Chen, J.; Buchkovich, M.L.; Mora, S.; et al. Discovery and refinement of loci associated with lipid levels. Nat. Genet. 2013, 45, 1274–1283. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Cases | Controls | p-Value | |
|---|---|---|---|
| Cohort | SCS/Sydney MAS | HCS | |
| Sample size (n) | 294 | 1105 | N/A |
| Age range (mean ± SD) | 95–106 (96.1 ± 4.1) | 55–65 (60.3 ± 2.8) | N/A |
| N (%) males | 90 (31) | 518 (47) | N/A |
| APOC3a C homozygotes, n (%) | 120 (40.1) | 475 (42.9) | 0.503564 |
| APOE ε4 carrier, n (%) | 43 (14.6) | 340 (30.8) | 1.49 × 10−8 |
| HDL (mean ± SD) (% missing) | 1.47 ± 0.45 (17.1) | 1.36 ± 0.37 (0.6) | 0.148557 |
| LDL (mean ± SD) (% missing) | 2.69 ± 1.02 (17.3) | 3.25 ± 0.91 (12.3) | 1.23 × 10−19 |
| TC (mean ± SD) (% missing) | 4.75 ± 1.16 (17.0) | 5.24 ± 1.03 (0) | 6.37 × 10−15 |
| TG (mean ± SD) (% missing) | 1.31 ± 0.63 (17.0) | 1.41 ± 1.12 (0.6) | 0.492286 |
| Phenotype [GWAS ref] | N SNPs p < 5 × 10−8 | N SNPs p < 5 × 10−5 | Total Number of GWAS SNPs Available |
|---|---|---|---|
| Exceptional longevity | |||
| Exceptional longevity [30] | 0 | 32 | 184,562 |
| Exceptional parental longevity [8] | 0 | 123 | 476,093 |
| Cardiovascular health | |||
| Myocardial infarction [31] | 35 | 107 | 19,607 |
| Stroke [32] | 11 | 226 | 471,632 |
| Coronary artery disease [33] | 96 | 447 | 460,589 |
| Essential hypertension (http://www.nealelab.is) | 2 | 107 | 476,069 |
| HDL [34] | 318 | 685 | 204,118 |
| LDL [34] | 301 | 652 | 202,316 |
| Cholesterol [34] | 367 | 797 | 204,123 |
| Triglycerides [34] | 238 | 592 | 201,879 |
| Odds Ratio (OR) | Standard Error [8] | p-Value | |
|---|---|---|---|
| PRSEL [30] | 1.20 | 0.068 | 0.00758 |
| PRSEPL [8] | 1.19 | 0.067 | 0.00804 |
| Sample Size (n) | Beta (β) | Standard Error [8] | p-Value | |
|---|---|---|---|---|
| PRSHDL | 1331 | 0.261 | 0.024 | 3.94 × 10−26 |
| PRSLDL | 1196 | 0.200 | 0.028 | 3.81 × 10−13 |
| PRSTC | 1336 | 0.176 | 0.026 | 1.66 × 10−11 |
| PRSTG | 1335 | 0.254 | 0.026 | 2.63 × 10−21 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Revelas, M.; Thalamuthu, A.; Oldmeadow, C.; Evans, T.-J.; Armstrong, N.J.; Riveros, C.; Kwok, J.B.; Schofield, P.R.; Brodaty, H.; Scott, R.J.; et al. Exceptional Longevity and Polygenic Risk for Cardiovascular Health. Genes 2019, 10, 227. https://doi.org/10.3390/genes10030227
Revelas M, Thalamuthu A, Oldmeadow C, Evans T-J, Armstrong NJ, Riveros C, Kwok JB, Schofield PR, Brodaty H, Scott RJ, et al. Exceptional Longevity and Polygenic Risk for Cardiovascular Health. Genes. 2019; 10(3):227. https://doi.org/10.3390/genes10030227
Chicago/Turabian StyleRevelas, Mary, Anbupalam Thalamuthu, Christopher Oldmeadow, Tiffany-Jane Evans, Nicola J. Armstrong, Carlos Riveros, John B. Kwok, Peter R. Schofield, Henry Brodaty, Rodney J. Scott, and et al. 2019. "Exceptional Longevity and Polygenic Risk for Cardiovascular Health" Genes 10, no. 3: 227. https://doi.org/10.3390/genes10030227