1. Introduction
Retroviruses have infiltrated vertebrate germline for millions of years by integrating as proviruses in host DNA, which have then passed down to the offspring through generations as inherited endogenous retroviruses (ERVs). The genomic ERV record represents retroviruses that were replicating at the time of integration and constitutes large fractions of contemporary vertebrate genomes, for example about 7–8% of human DNA [
1,
2] and about 3% of the chicken genome [
3]. The genomic ERV record thus presents a remarkable source for an evolutionary perspective on the biology and interactions among retroviruses and their hosts.
Diverse sets of ERVs can be identified across all studied vertebrate genome assemblies [
4] by screening for structural hallmarks including long terminal repeats (LTRs), which flank the ERV
gag, pol, and
env genes [
5]. Over time, ERV loci may become fixed in the host population, either due to genetic drift of those loci that are least harmful or due to selection on beneficial insertions [
2]. ERV contributions to the host genome structure and function include providing a substrate for genomic recombination, and effects on the host transcriptome resulting from their integration and expression with diverse effects on host genome function and evolution. Among positive effects are the expression of viral gene products as useful new genes in the host [
6], modification of chromosomal gene expression by ERVs including promoter, enhancer, and insulator functions, as well as alternative splice signals from ERV integrations in host transcription units or adjacent to chromosomal genes [
2,
7]. On the other hand, is the potential for host gene disruption, as well as the potential for somatic spread of replicating retroviruses leading to pathogenic consequences for the host [
2,
8,
9]. ERV-mediated genomic recombination can further contribute to the organization and plasticity of the host’s genome [
10,
11,
12,
13]. Overall, it is plausible that ERVs have had considerable effects on host genome function and evolution across the entire vertebrate lineage, by shuffling genomic regions, exons, and regulatory genetic sequences into new contexts and thereby altering the dynamic functions of the host DNA.
It is desirable to identify orthologous ERV loci across the compared host lineages in order to evaluate potential effects of retroviruses and ERVs on host biology because it allows for connecting ERV integrations to host phenotypic differences and evolutionary history. ERV studies have benefited from recent advancements in sequencing technology and a growing catalogue of reference host genome assemblies, where much focus has been placed on comparing ERV records across related host species reference genome assemblies, an approach that suffers from undersampling of the diversity within vertebrate species, and thus presents challenges for reaching a better understanding of potential factors that contribute to the long-term retrovirus-host associations [
4]. More recently, studies utilizing re-sequencing data to target searches for integration differences among selected ERVs within a host population to explain activities during recent evolution have made efforts to address this issue for specific virus types in host populations [
14,
15].
In an attempt to further explore ERV-host associations in a hitherto un-examined system, we make use of an artificial selection system where selection lines of domestic chicken that have been undergoing strong bi-directional selection on body weight at eight weeks of age for more than 50 years [
16]. This selection-scheme, from a single founder population, has generated extreme phenotypes with more than 10-fold difference in average weights between the two chicken lines. We utilized whole-genome re-sequencing data from these chicken lines, as well as an outgroup commercial chicken line, to investigate ERV insertion variation and potential evolutionary contributions from inherited ERVs on host genome function.
Domestic animals provide rare possibilities, currently not feasible in human biomedicine, to study connections between genes, phenotypes, and biological function [
17]. Crossbreeding of domestic animals is also a useful tool to determine genomic differences, making it possible to apply genetic analyses to reveal loci controlling phenotypic traits that have been selected during domestication [
18].
The rationale for utilizing chicken as a model dates back more than 100 years to pioneering studies of retroviruses and ERVs, reviewed in [
19,
20], and the availability of sequence data from the chicken selection pedigree established in 1957 (see above), which, measured by the response in phenotypic traits and single nucleotide polymorphism (SNP) allele-frequency divergence, has accumulated changes that have been estimated by Johansson et al. to require about 5000 years to evolve in natural populations [
21]. Overall, the chicken selection lines present a promising model for identifying ERV divergence and interpreting observations in the context of previously known results in this system, thereby estimating ERV contributions to dynamics of complex genetic traits of their hosts.
Here, we identify ERV insertion differences across available re-sequenced genomes derived from the two bi-directionally growth-selected chicken lines and compare candidate ERV loci with a commercial layer chicken outgroup. We map insertions and deletions to establish their positions relative to the adjacent host genomic landscape and compare candidate loci associated by sequence similarity to ERVs identified in the Red junglefowl reference assembly (version galGal3) along with reference retroviral sequences within a phylogenetic framework.
3. Results
Whole-genomes from domestic High-Growth (H), Low-Growth (L), and White Leghorn (W) chicken selection lines were previously sequenced using high throughput SOLiD technology and mapped to the chicken reference assembly (version galGal3) by Rubin et al. [
18], see
Table 1. Here, we utilized the RetroTector software [
5] to identify ERVs in the Red junglefowl (version galGal3) assembly, which were used as an independent sequence library to map ERV-associated reads from the re-sequenced chicken lines that could then be mated with their respective chromosomal mapping reads for locating ERV-host DNA insertion junctions, even in cases where the insertion was absent from the reference assembly. Up- and downstream ERV-host junctions were clustered and paired using stringency criteria tuned for identifying about 10,000 loci, the expected number of ERV and solo LTR loci based on RetroTector results and a previously estimated 1:20 ratio between complete ERVs and solo LTRs, which are generated by homologous recombination between the two proviral LTRs [
1]. The clustering of paired sequence reads identified 12,709 candidate ERV loci, of which 8340 candidate loci indicated distinct differences, measured as absence or near-absence of ERV-associated reads in at least one of the three compared chicken lines. Bonferroni correction for multiple testing left 369 differentiated candidate ERV loci. Among these candidate loci, 115 ERVs were adjacent to, or located within, 229 host genes considering 150 kb distances up- and downstream of the candidate loci, see
Table 2 and
Supplementary Information Table S1.
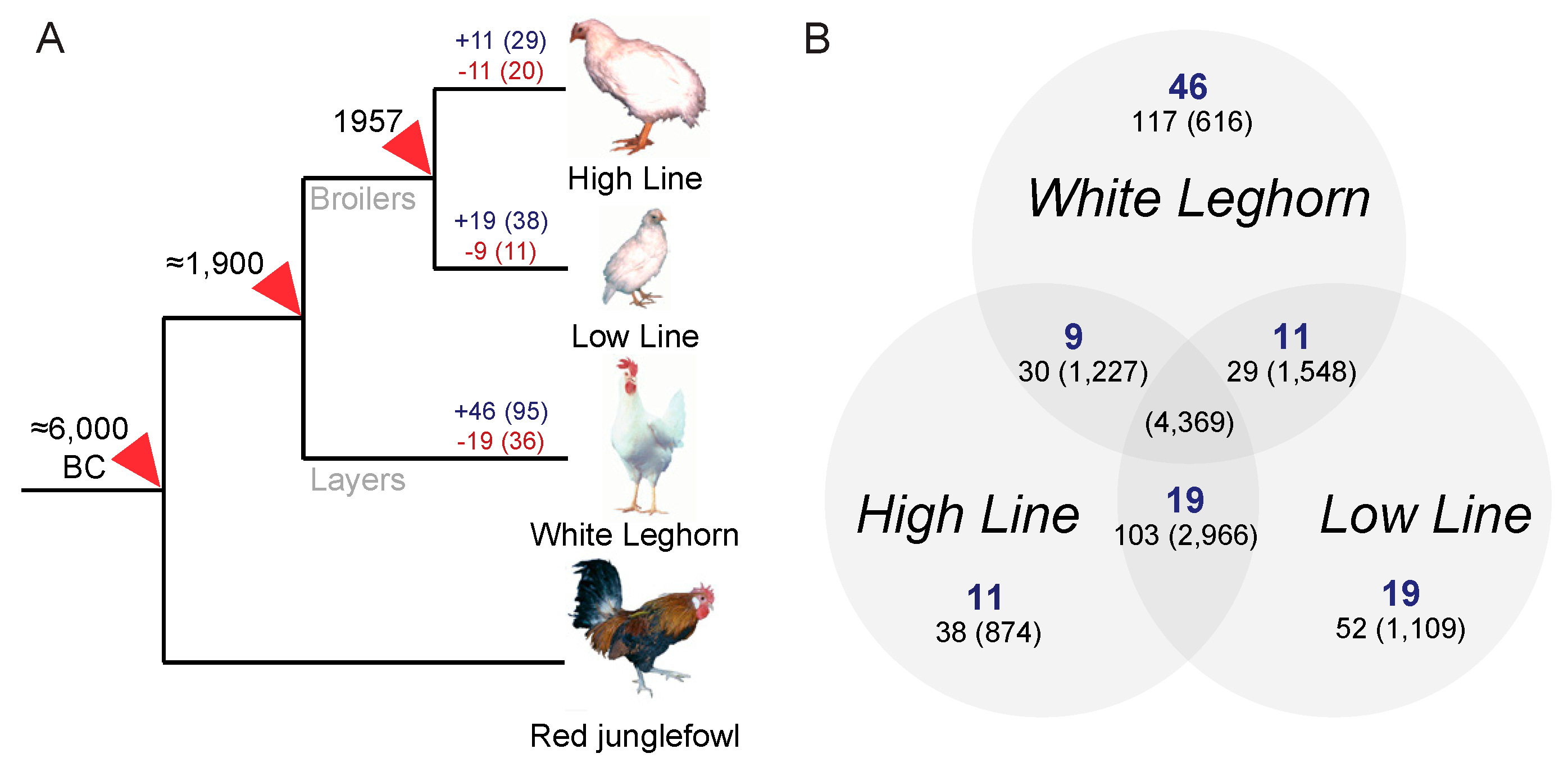
The bi-directionally growth-selected chicken lines (H and L) diverged from a single broiler founder population about 60 years ago and were separated more than 100 years ago from the branch leading to the comparison outgroup represented here by the commercial White Leghorn (layer) chicken. For reference, the compared chicken lines share a relatively recent common ancestry, compared to the reference genome assembly, generated from the Red junglefowl,
Gallus gallus, which was separated from the investigated chicken lines about 8000 years ago when chicken was first domesticated, see
Figure 1. However, even this split is recent compared to datasets that have been the subject of previous studies [
4,
14,
15,
26], and thus the use of ERV loci comparison in a small host pedigree, such as the domestic chicken lines, relies on that integration differences may be observed as a result from selection during domestication that could require many thousands of years to become fixed in wild host populations [
21]. The observed branch-specific ERV loci differences across the domestic chicken selection lines broadly reflects the time scale after divergence as the growth-selected broiler chicken lines (H and L) were separated about half of the time since the layer outgroup (W) separated from the domesticated broiler chickens, see
Figure 1, indicating that differences in ERV makeups may provide potential traceable markers for host evolution, see
Figure 1,
Table 2.
To explore potential connections between the observed divergent ERV loci across the domestic chicken lines, we intersected chromosomal positions with the reference assembly host genes (version galGal3, downloaded from the UCSC genome browser,
http://genome.ucsc.edu) and previously determined selective sweeps for the H and L chicken selection lines [
18,
21]. Although some ERVs overlapped with domestication sweep regions, see
Supplementary Information Table S1, the observed overlap did not deviate significantly from the expectation (
p = 0.1, binomial test), given the size of the sweep areas and the number of detected ERV insertions elsewhere in the host genomes. In addition, gene ontology searches were inconclusive and could not establish links between ERVs and adjacent host genes that could help explain the distinct phenotypes. We, therefore, analyzed candidate ERV insertion orientations and distances relative to host genes. Candidate ERV loci within host gene transcripts show a clear bias in antisense orientation relative to the host gene transcript, which could be explained by purifying selection due to potential splice interference from canonical splice signals as previously discussed [
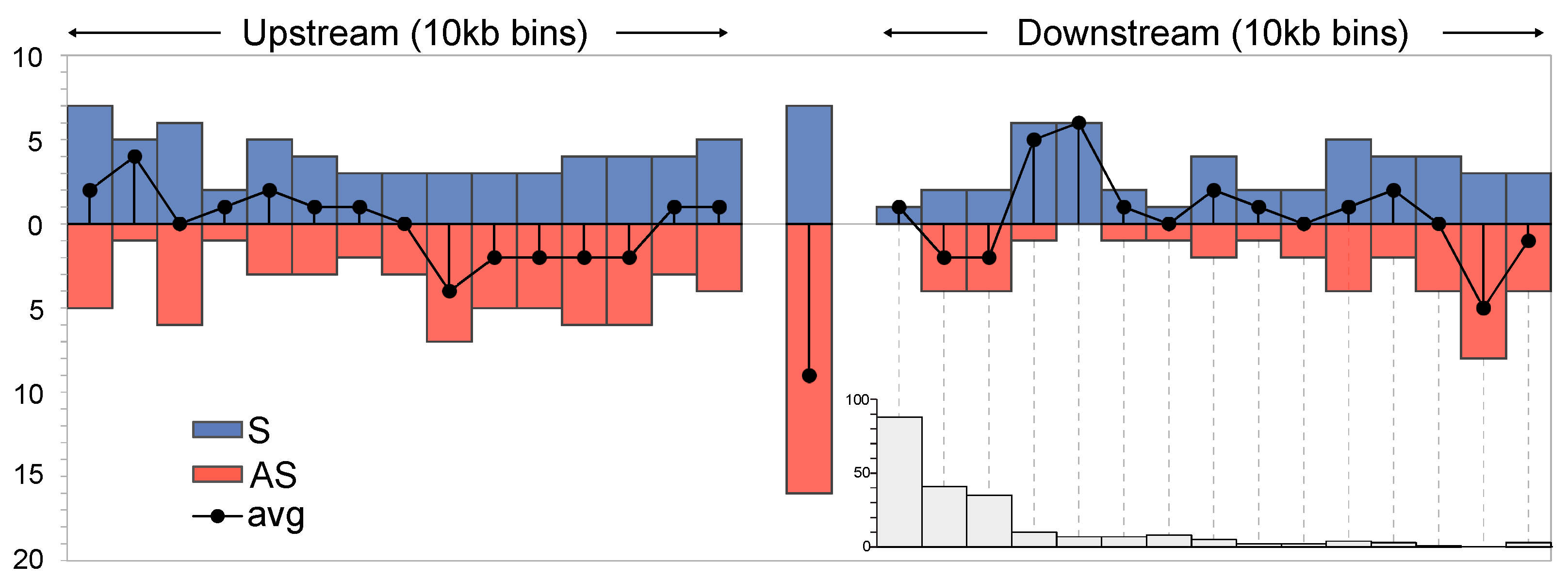
28]. Intergenic ERV orientations relative to host genes fluctuate up- and downstream and a bias pattern is not clear given the limited data, see
Figure 2. It thus appears that intergenic ERV insertions may not influence host genome function to the same extent as intragenic ERVs.
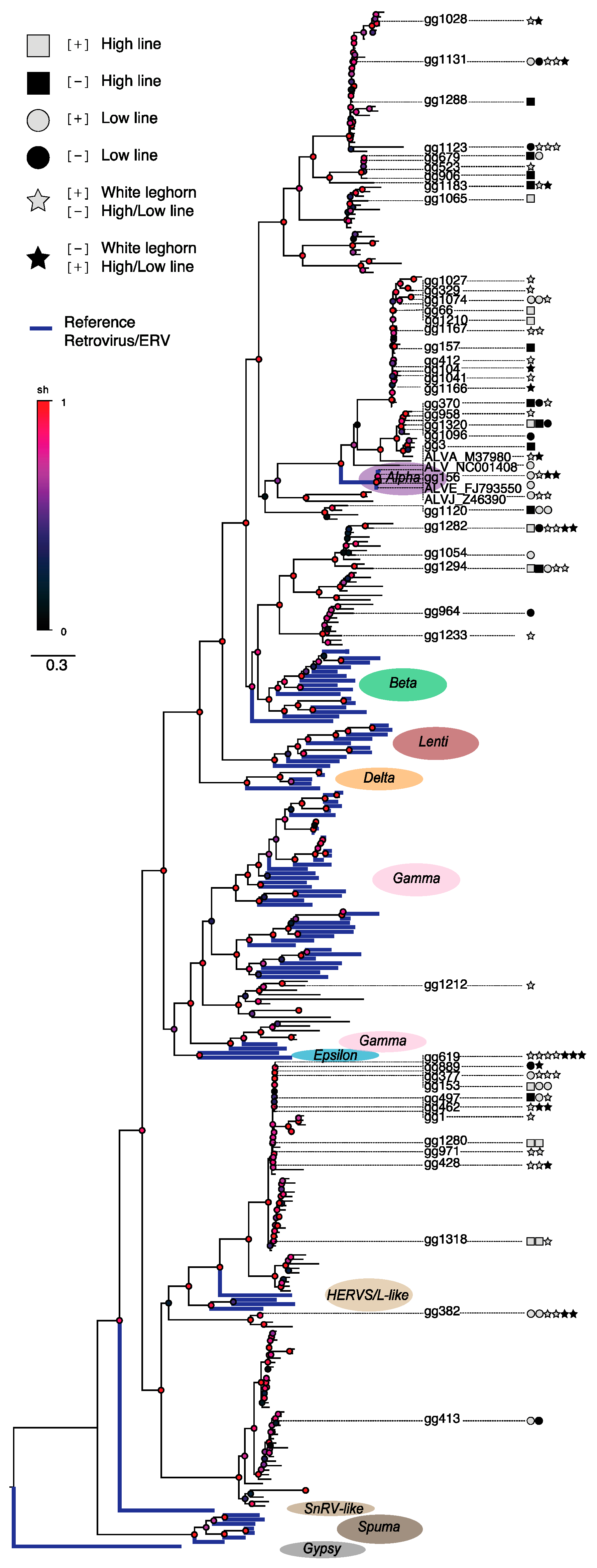
To investigate relationships between the observed ERV loci varying across the analyzed chicken lines, we constructed a phylogenetic tree based on ERVs identified by the RetroTector software [
5] in the Red junglefowl reference assembly (version galGal3) and appended reference retroviral sequences for comparisons as previously described [
4,
25,
26]. Since the insert sizes and read lengths of ERV-associated mate-pair reads only allow limited coverage into the candidate ERV loci, it is useful to align reads to reference assembly ERVs that could build a phylogenetic framework, and from which the best ERV match for candidate ERV loci by can be determined, see
Figure 3.
In agreement with the observed lack of significant associations between candidate ERV loci and adjacent host genes (see above), divergent ERV loci in the domestic H, L, and W chicken lines located across the phylogenetic tree that was rooted on a distant outgroup, rather than being found inside any specific retroviral clade, which is what could be expected if variation was due to retroviral expansion after the last common ancestor. Instead, the result indicates that the observed candidate ERV insertion differences do not result from recent retrovirus replication and integrations as ERVs in one or two of the chicken lineages, but rather it is consistent with standing variation of segregating ERV loci present at the onset of the bi-directional selection experiment as well as during breed formation since the domestication of chicken. Multiple radiations involving candidate ERV loci associated with assembly ERVs showing short terminal branch lengths indicate relatively recent expansions occurring within several retroviral genera across the phylogeny. It seems plausible that these radiations have generated a substantial number of segregating ERV insertions in the domestic chicken lines, thus providing the standing variation that explains the observed differences in ERV makeups, and that the number of divergent ERV loci is largely a product of the accelerated genomic divergence caused by the strong selection imposed on the H and L lines specifically, as well as directed selection of host features during domestication, which has affected all three studied (H, L, and W) chicken lines.
4. Discussion
The known breeding history and well-studied phenotypic traits among domestic animals make them first-rate model organisms to identify potential ERV contributions to biological functions and dynamics of complex genetic traits. Rare genomic changes resulting in host phenotypes that would require thousands of years to establish, or become lost, in wild host populations may be selected for in domestic settings during fewer generations [
17,
21,
29]. This type of genomic data presents an excellent chance to study differences in genomic ERV makeups across many chicken selection lines.
Here, we utilized whole-genome sequences from two bi-directionally growth-selected domestic broiler chicken lines and a distantly related domestic layer chicken line [
18] for identifying and comparing candidate ERV insertion differences. Using an independent ERV search library, it is possible to identify non-reference assembly ERV in the different host lineages. We show that the domestic chicken carries a large number of segregating ERVs, evidenced by the observation that 65% of detected loci display a nominal difference in frequencies and more than 350 insertions are significantly differentiated. Standing variation has previously been shown to contribute to the majority of the alleles under selection in the H and L lines [
21,
22,
29] and here we show that segregating ERVs are a part of this variation, and thus they form part of the potential substrate for selection in these, and other, chicken lines.
As high-throughput parallel sequencing technologies generate short reads and limited coverage into the ERV loci depending on mate-pair insert sizes for reliable chromosomal anchoring, we utilized ERVs identified in the Red junglefowl reference assembly to generate an independent ERV search library and anchored loci to chromosomal positions by ERV-associated short reads mate-pair mapping. Despite limited ERV sequence coverage, it is thus possible to associate the best fit for candidate ERV loci reads with assembly ERV sequences, which could be used to construct phylogenetic frameworks and to determine associations between ERV loci and host genomic landscapes.
The whole-genome sequences were generated from pooled individual DNA, see
Table 1, which complicates assessments of ERV presence/absence and we, therefore, used a conservative approach by considering loci where one or two of the three domestic chicken lines indicated missing or present ERV-associated reads. Given the large phenotypic differences between the high- and low-growth chicken lines, the domestic animal model presents a promising system to determine potential influences from ERVs on host genome structure and function.
However, although gene ontology searches for genes adjacent to divergent candidate ERV loci could not explain host phenotypic variation, which could be due to the known highly polygenic nature of the trait under selection [
21,
29], intersection of ERV loci positions with previously determined domestication sweep signals [
18,
21,
29] showed only a weak association. Together, the results suggest that pre-existing ERV variation derived from a common host ancestor segregate in the domestic chicken selection lines today. This notion is supported by the estimated age range of loci that are divergent loci between the domestic chicken lines, which include loci that are presumably old, based on sequence similarities to re-constructed reference assembly ERVs, as well as newer reference assembly ERV insertions, indicating that the divergence represents a frequency shift among ERV loci that segregate in the host population.
Similar observations have also recently been made in other vertebrate host populations [
26], demonstrating limitations when assessing historic retrovirus activities from the genomic ERV record using reference assemblies compared to host population data [
30], due to the severe sampling effect and associated loss of diversity introduced by reducing a species population down to a single reference genome.
While it can be informative to study ERV variation from host species assemblies covering multiple species over long evolutionary time scales [
4,
25], analyses along single species phylogenies provide additional information regarding ERV variation and expression [
31]. To analyze more recent ERV activities, it has also been successful to employ targeted analysis of specific ERVs in single host species population data [
14,
15]. However, sampling constraints complicate identification of standing variations in ERV makeups in these systems, and the broad searches in controlled genome groups that the domestic animal selection pedigrees provide are not easily achieved under such conditions. By narrowing the time scale using domestic and wild animal pedigrees, it has been possible to estimate segregating ERV variation for a broad range of ERV clades in host populations [
26]. Use of PCR to investigate polymorphisms and incomplete lineage sorting was recently demonstrated for young ERVs [
32], and further refining these types of studies by analyzing the recently diverged growth-selected chicken pedigree in this study, we conclude that standing ERV variation is a common feature in contemporary vertebrate populations.
In summary, it appears increasingly important to employ careful experimental design to control the occurrence of artifacts and incorrect inferences due to unbalanced sampling in analyses aimed at evaluating host species ERV makeups. In order to obtain valid comparisons from population and distantly related genomes, it is valuable to focus on well-known pedigrees like those offered by domestic animal selection lines, where the prior knowledge makes it possible to compare observed patterns with expectations that are based on the evolutionary context of the specific case with higher precision than is generally achievable in natural populations. Sequencing and analyses of domestic animal populations and single genomes from known selection pedigrees facilitated by improved sequencing technologies that provide depth and coverage over long insertion sizes together with newly developed and fine-tuned analysis methods will facilitate mapping of ERVs previously not feasible and thereby generate new knowledge about contributions from retroviruses and ERVs to host genome function and evolution.






{kind=link}
{kind=link}
{kind=link}