Investigating the DNA-Binding Site for VirB, a Key Transcriptional Regulator of Shigella Virulence Genes, Using an In Vivo Binding Tool

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Bacterial Strains, Plasmids, and Media
2.2. Construction of the In Vivo Binding Tool, pBT-Empty
2.3. Construction of the In Vivo Binding Tool Derivatives
2.4. Purification of VirB
2.5. DNase I Protection Assays
2.6. β-Galactosidase Assays
3. Results
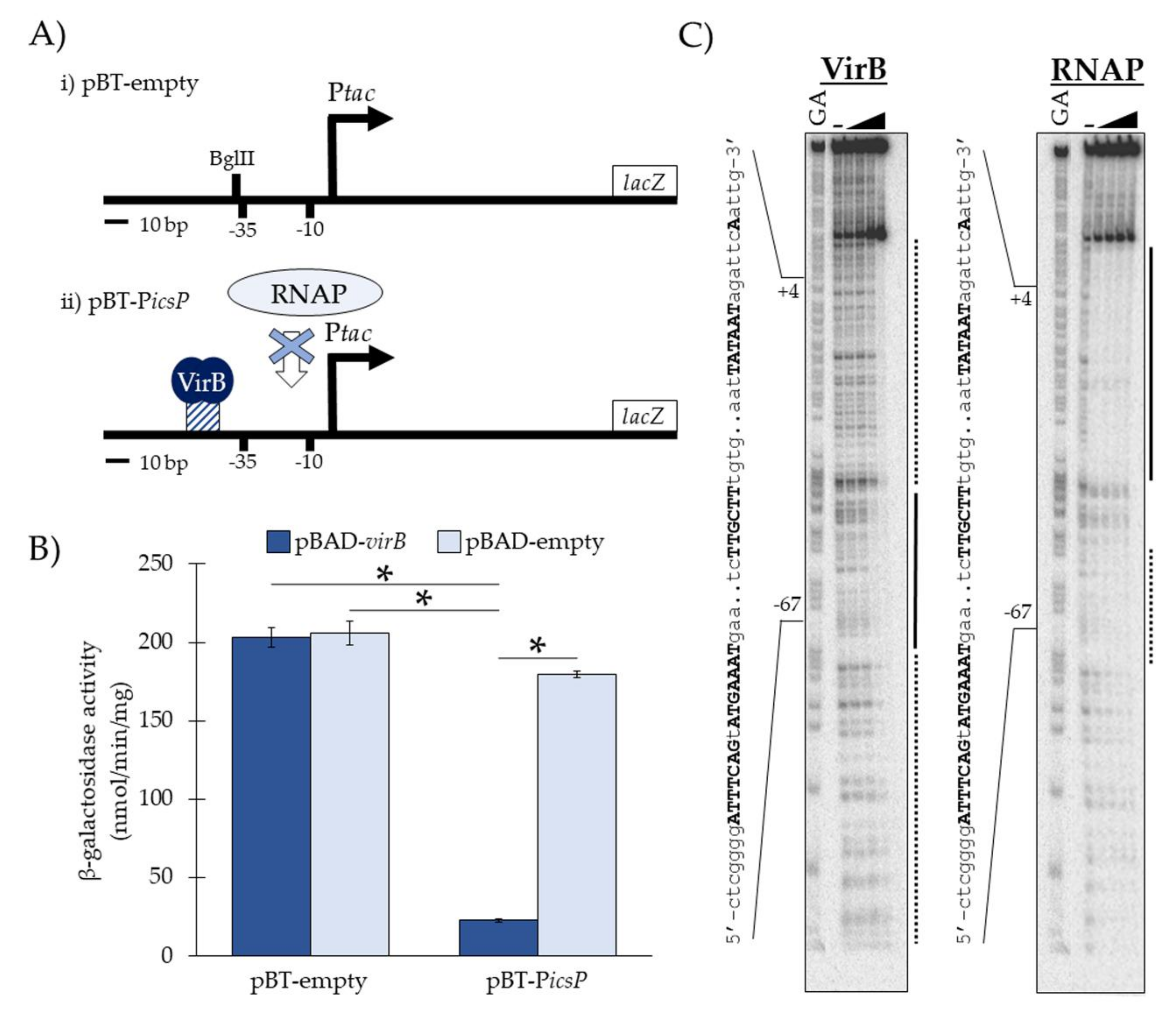
3.1. Experimental Validation of the In Vivo Binding Tool
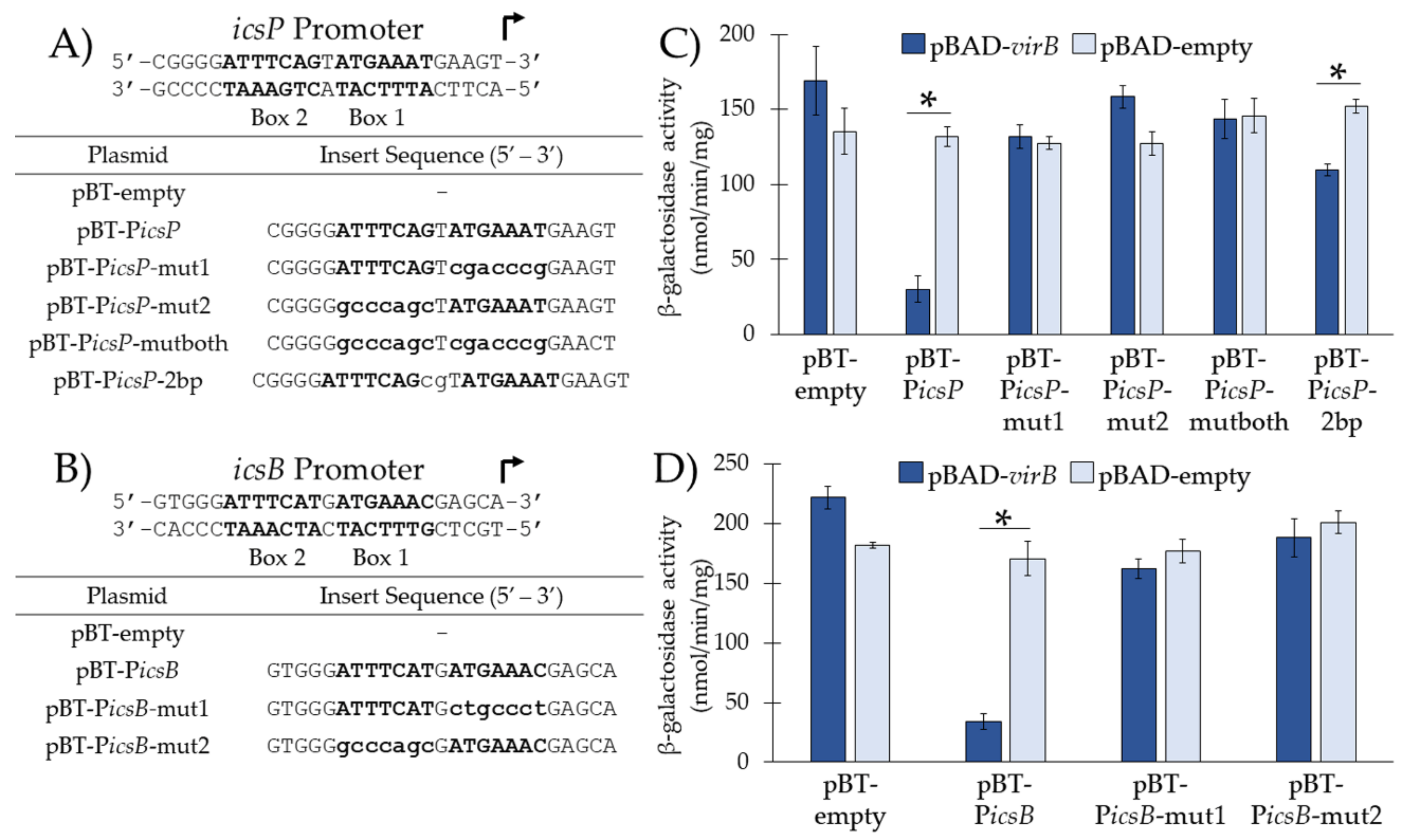
3.2. VirB Binds the Near-Perfect Inverted Repeats from the icsP and icsB Promoters In Vivo
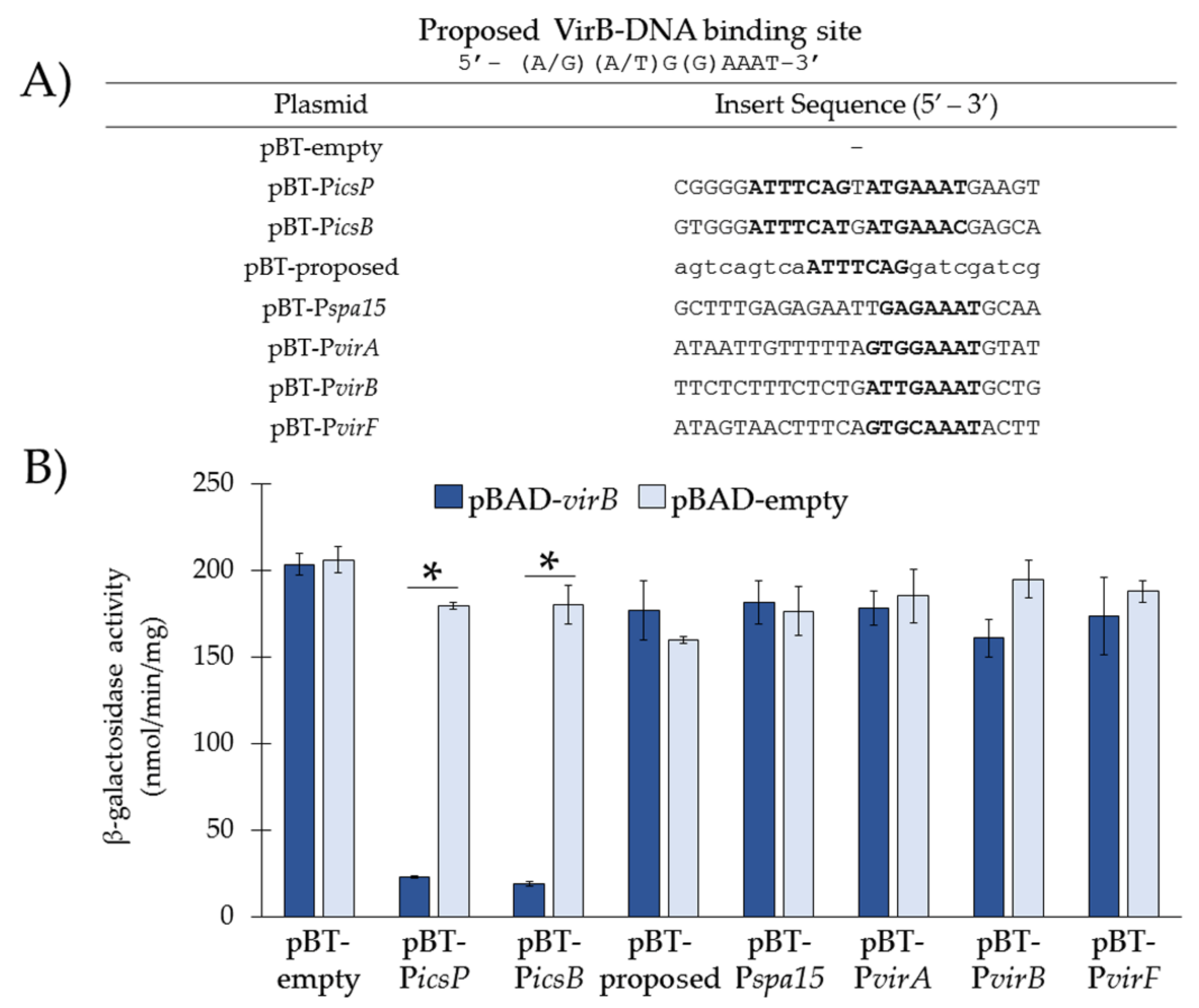
3.3. Re-Examination of VirB-Binding Sites Described in the Literature Using our In Vivo Binding Tool
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Adler, B.; Sasakawa, C.; Tobe, T.; Makino, S.; Komatsu, K.; Yoshikawa, M. A dual transcriptional activation system for the 230kb plasmid genes coding for virulence-associated antigens of Shigella flexneri. Mol. Microbiol. 1989, 3, 627–635. [Google Scholar] [CrossRef] [PubMed]
- Maurelli, A.T.; Blackmon, B.; Curtiss, R., 3rd. Temperature-dependent expression of virulence genes in Shigella species. Infect. Immun. 1984, 43, 195–201. [Google Scholar] [PubMed]
- Porter, M.E.; Dorman, C.J. Differential regulation of the plasmid-encoded genes in the Shigella flexneri virulence regulon. Mol. Gen. Genet. 1997, 256, 93–103. [Google Scholar] [CrossRef] [PubMed]
- Basta, D.W.; Pew, K.L.; Immak, J.A.; Park, H.S.; Picker, M.A.; Wigley, A.F.; Hensley, C.T.; Pearson, J.S.; Hartland, E.L.; Wing, H.J. Characterization of the ospZ promoter in Shigella flexneri and its regulation by VirB and H-NS. J. Bacteriol. 2013, 195, 2562–2572. [Google Scholar] [CrossRef] [PubMed]
- Beloin, C.; Dorman, C.J. An extended role for the nucleoid structuring protein H-NS in the virulence gene regulatory cascade of Shigella flexneri. Mol. Microbiol 2003, 47, 825–838. [Google Scholar] [CrossRef] [PubMed]
- Turner, E.C.; Dorman, C.J. H-NS antagonism in Shigella flexneri by VirB, a virulence gene transcription regulator that is closely related to plasmid partition factors. J. Bacteriol. 2007, 189, 3403–3413. [Google Scholar] [CrossRef] [PubMed]
- Weatherspoon-Griffin, N.; Picker, M.A.; Pew, K.L.; Park, H.S.; Ginete, D.R.; Karney, M.MA.; Usufzy, P.; Castellanos, M.I.; Duhart, J.C.; Harrison, D.J.; et al. Insights into transcriptional silencing and anti-silencing in Shigella flexneri: A detailed molecular analysis of the icsP virulence locus. Mol. Microbiol. 2018, 108, 505–518. [Google Scholar] [CrossRef]
- Wing, H.J.; Yan, A.W.; Goldman, S.R.; Goldberg, M.B. Regulation of IcsP, the Outer Membrane Protease of the Shigella Actin Tail Assembly Protein IcsA, by Virulence Plasmid Regulators VirF and VirB. J. Bacteriol. 2004, 186, 699–705. [Google Scholar] [CrossRef] [PubMed]
- Taniya, T.; Mitobe, J.; Nakayama, S.; Mingshan, Q.; Okuda, K.; Watanabe, H. Determination of the InvE binding site required for expression of IpaB of Shigella sonnei virulence plasmid: Involvement of a ParB BoxA-like sequence. J. Bacteriol. 2003, 185, 5158–5165. [Google Scholar] [CrossRef] [PubMed]
- Castellanos, M.I.; Harrison, D.J.; Smith, J.M.; Labahn, S.K.; Levy, K.M.; Wing, H.J. VirB Alleviates H-NS Repression of the icsP Promoter in Shigella flexneri from Sites More Than One Kilobase Upstream of the Transcription Start Site. J. Bacteriol. 2009, 191, 4047–4050. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, H.; Arakawa, E.; Ito, K.; Kato, J.; Nakamura, A. Genetic analysis of an invasion region by use of a Tn3-lac transposon and identification of a second positive regulator gene, invE, for cell invasion of Shigella sonnei: Significant homology of InvE with ParB of plasmid P1. J. Bacteriol. 1990, 172, 619–629. [Google Scholar] [CrossRef] [PubMed]
- McKenna, S.; Beloin, C.; Dorman, C.J. In vitro DNA-binding properties of VirB, the Shigella flexneri virulence regulatory protein. FEBS Lett. 2003, 545, 183–187. [Google Scholar] [CrossRef]
- Beloin, C.; McKenna, S.; Dorman, C.J. Molecular Dissection of VirB, a Key Regulator of the Virulence Cascade of Shigella flexneri. J. Bacteriol. 2002, 277, 15333–15344. [Google Scholar]
- Williams, S.M.; Wing, H.J.; Busby, S.J.W. Repression of transcription initiation by Escherichia coli FNR protein: Repression by FNR can be simple. FEMS Microbiol. Lett. 1998, 163, 203–208. [Google Scholar] [CrossRef] [PubMed]
- de Boer, H.A.; Comstock, L.J.; Vasser, M. The tac promoter: A functional hybrid derived from the trp and lac promoters. Proc. Natl. Acad. Sci. 1983, 80, 21–25. [Google Scholar] [CrossRef] [PubMed]
- LB (Luria-Bertani) liquid medium. Cold Spring Harb. Protoc. 2006, 2006, pdb.rec8141. [CrossRef]
- Chang, A.C.; Cohen, S.N. Construction and characterization of amplifiable multicopy DNA cloning vehicles derived from the P15A cryptic miniplasmid. J. Bacteriol. 1978, 134, 1141–1156. [Google Scholar] [PubMed]
- Farinha, M.A.; Kropinski, A.M. Construction of Broad-Host-Range Plasmid Vectors for Easily Visible Selection and Analysis of Promoters. J. Bacteriol. 1990, 172, 3496–3499. [Google Scholar] [CrossRef]
- Kane, K.A.; Dorman, C.J. VirB-Mediated Positive Feedback Control of the Virulence Gene Regulatory Cascade of Shigella flexneri. J. Bacteriol. 2012, 194, 5264–5273. [Google Scholar] [CrossRef]
- Bahl, C.P.; Wu, R.; Stawinsky, J.; Narang, S.A. Minimal length of the lactose operator sequence for the specific recognition by the lactose repressor. Proc. Natl. Acad. Sci. 1977, 74, 966–970. [Google Scholar] [CrossRef]
- Maxam, A.M.; Gilbert, W. A method for determining DNA sequence by labeling the end of the molecular and cleaving at the base. Isolation of DNA fragments, end-labeling, cleavage, electrophoresis in polyacrylamide gel and analysis of results. Mol. Biol. 1986, 20, 581–638. [Google Scholar]
- Miller, J. Experiments in Molecular Genetics. Cold Spring Habor Laboratory Press: Cold Spring Harbor, NY, USA, 1972. [Google Scholar]
- Jin, Q.; Yuan, Z.; Xu, J.; Wang, Y.; Shen, Y.; Lu, W.; Wang, J.; Liu, H.; Yang, J.; Yang, F.; et al. Genome sequence of Shigella flexneri 2a: Insights into pathogenicity through comparison with genomes of Escherichia coli K12 and O157. Nucleic Acids Res. 2002, 30, 4432–4441. [Google Scholar] [CrossRef] [PubMed]
- Le Gall, T.; Mavris, M.; Martino, M.C.; Bernardini, M.L.; Denamur, E.; Parsot, C. Analysis of virulence plasmid gene expression defines three classes of effectors in the type III secretion system of Shigella flexneri. Microbiology 2005, 151, 951–962. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Zou, T.; Mu, Z.; Qin, B.; Yang, J.; Waltersperger, S.; Wang, M.; Cui, S.; Jin, Q. Structural insights into VirB-DNA complexes reveal mechanism of transcriptional activation of virulence genes. Nucleic Acids Res. 2013, 41, 10529–10541. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Bacterial Strain | Description | Source |
|---|---|---|
| AWY3 | Shigella flexneri serotype 2a strain 2457T virB::Tn5; Knr. | [8] |
| JM109(DE3) | Escherichia coli strain derived from JM109. | Promega |
| Plasmids | Description | Source |
| pACYC177 | Low-copy number plasmid vector with a p15A ori; Ampr. | [17] |
| pQF50 | Promoterless lacZ transcriptional fusion vector; Cbr. | [18] |
| pQE-60 | Vector for C-terminally fused His6-tagged insert; Ampr. | Qiagen |
| pBlueScript II KS(+) | Multi-copy cloning vector; Ampr. | Stratagene |
| pHJW14 | pBAD18; Cmr. | [8] |
| pHJW16 | pHJW14-virB; Cmr. | [8] |
| pHJW43 | Source of gfp used to create pBT-empty; Ampr. | This work |
| pAJH03 | IPTG inducible plasmid carrying His6-tagged virB. | [7] |
| pNWG08 | pBlueScript II KS(+) with NotI BamHI sequence of pBT-PicsP; Ampr. | This work |
| pBT-empty | pACYC177 carrying Ptac-lacZ with a unique BglII site immediately adjacent to the −35 promoter element. | This work |
| pBT-PicsP | pBT-empty with PicsP Box 1 and 2 separated by a single nucleotide spacer 5′-ATTTCAGtATGAAAT-3′ [10] | This work |
| pBT-PicsP-mut1 | pBT-empty with mutated PicsP Box 1 | This work |
| pBT-PicsP-mut2 | pBT-empty with mutated PicsP Box 2 | This work |
| pBT-PicsP-mutboth | pBT-empty with mutated PicsP Box 1 and Box 2 | This work |
| pBT-PicsP-2bp | pBT-empty with PicsP Box 1 and 2 with an added 2 bp spacer | This work |
| pBT-PicsB | pBT-empty with PicsB Box 1 and 2 separated by a single nucleotide spacer 5′-ATTTCATgATGAAAC-3′ [6] | This work |
| pBT-PicsB-mut1 | pBT-empty with mutated PicsB Box 1 | This work |
| pBT-PicsB-mut2 | pBT-empty with mutated PicsB Box 2 | This work |
| pBT-proposed | pBT-empty with proposed 7 bp VirB-binding site [9] flanked by scrambled sequences | This work |
| pBT-Pspa15 | pBT-empty with putative VirB-binding site 5′-GAGAAAT-3′ [9] from Pspa15 | This work |
| pBT-PvirA | pBT-empty with putative VirB-binding site 5′-GTGGAAAT-3′ [9] from PvirA | This work |
| pBT-PvirB | pBT-empty with putative VirB-binding site 5′-ATTGAAAT-3′ [19] from PvirB | This work |
| pBT-PvirF | pBT-empty with putative VirB-binding site 5′-GTGCAAAT-3′ [19] from PvirF | This work |
| Primer1 | Sequence (5′ to 3′) | Description or Use |
|---|---|---|
| W342 | ctagaggatccccgggtacccg | Amplification of lacZ from pQF50 |
| W343 | gcattactcgagttccttacgcgaaagacgggc | |
| W344 | ggaaactagtcgattcgtaggccttgctttgtgagcggataac | Amplification of Ptac from pQE60 |
| W345 | cgcggtcgaccccatatcaccagctcaccg | |
| W395 | ctagaactagtcgattcgtagatcttgctttgtgagcgg | Mutagenic primers to generate unique BglII site required for construction of pBT-empty |
| W396 | agctagaagcttctagagatcccc | |
| W412 | tgcattgcggccgcttttctgtcagtggagaggg | Amplification of ‘gfp required for construction of pBT-empty |
| W413 | actgcagccggcaagaaggaccatgtgg | |
| W414 | gatctCGGGGATTTCAGTATGAAATGAAGTg | Annealed to generate wild-type PicsP Box 1 & 2 |
| W463 | gatccACTTCATTTCATACTGAAATCCCCGa | |
| W527 | gatctCGGGGATTTCAGTcgacccgGAAGTg | Annealed to generate mutated PicsP Box 1 |
| W528 | gatccACTTCcgggtcgACTGAAATCCCCGa | |
| W529 | gatctCGGGGgcccagcTATGAAATGAAGTg | Annealed to generate mutated PicsP Box 2 |
| W530 | gatccACTTCATTTCATAgctgggcCCCCGa | |
| W416 | gatctCGGGGgcccagcTcgacccgGAACTg | Annealed to generate mutated PicsP Box 1 & 2 |
| W464 | gatccAGTTCcgggtcgAgctgggcCCCCGa | |
| W587 | gatctCGGGGATTTCAGcgTATGAAATGAAGTg | Annealed to create insert with 2 bp between PicsP Box 1 & 2 |
| W588 | gatccACTTCATTTCATacGCTGAAATCCCCGa | |
| W443 | gatctTGCTCGTTTCATCATGAAATCCCACg | Annealed to generate wild-type PicsB Box 1 & 2 |
| W444 | gatccGTGGGATTTCATGATGAAACGAGCAa | |
| W445 | gatctTGCTCagggcagCATGAAATCCCACg | Annealed to generate mutated PicsB Box 1 |
| W446 | gatccGTGGGATTTCATGctgccctGAGCAa | |
| W474 | gatctTGCTCGTTTCATCgctgggcCCCACg | Annealed to generate mutated PicsB Box 2 |
| W450 | gatccGTGGGgcccagcGATGAAACGAGCAa | |
| W596 | gatctagtcagtcaATTTCAGgatcgatgcc | Annealed to generate proposed VirB-binding site |
| W597 | gatccCGATCGATCCTGAAATTGACTGACTa | |
| W627 | gatctGCTTTGAGAGAATTGAGAAATGCAAg | Annealed to generate putative VirB-binding site at Pspa15 |
| W628 | gatccTTGCATTTCTCAATTCTCTCAAAGCa | |
| W629 | gatctATAATTGTTTTTAGTGGAAATGTATg | Annealed to generate putative VirB-binding site at PvirA |
| W630 | gatccATACATTTCCACTAAAAACAATTATa | |
| W631 | gatctTTCTCTTTCTCTGATTGAAATGCTGg | Annealed to generate putative VirB-binding site at PvirB |
| W632 | gatccCAGCATTTCAATCAGAGAAAGAGAAa | |
| W633 | gatctATAGTAACTTTCAGTGCAAATACTTg | Annealed to generate putative VirB-binding site at PvirF |
| W634 | gatccAAGTATTTGCACTGAAAGGGACTATa | |
| W119 | gccagggttttgggagtcacga | Sequencing primer, M13F |
| W120 | gagcggataacaatttcacacagg | Sequencing primer, M13R |
| W482 | tgtggtgcaacgggcgctgg | Sequencing primer |
| W483 | agaagcctgcgatgtcggtt | Sequencing primer |
| W484 | caccgatattatttgcccga | Sequencing primer |
| W485 | cctctggatgtcgctccaca | Sequencing primer |
| W486 | ggcagcatcaggggaaaacc | Sequencing primer |
| W487 | gcacatttccccgaaaagtg | Sequencing primer |
| W488 | ggaagacgtacggggtatac | Sequencing primer |
| W489 | ccagctcgatgcaaaaatcc | Sequencing primer |
| W490 | ccacccagtcccagacgaag | Sequencing primer |
| W491 | ccacagcggatggttcggat | Sequencing primer |
| W492 | cggtttatgcagcaacgaga | Sequencing primer |
| W541 | gcggccgctctagaactagtcg | Sequencing primer |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Karney, M.M.A.; McKenna, J.A.; Weatherspoon-Griffin, N.; Karabachev, A.D.; Millar, M.E.; Potocek, E.A.; Wing, H.J. Investigating the DNA-Binding Site for VirB, a Key Transcriptional Regulator of Shigella Virulence Genes, Using an In Vivo Binding Tool. Genes 2019, 10, 149. https://doi.org/10.3390/genes10020149
Karney MMA, McKenna JA, Weatherspoon-Griffin N, Karabachev AD, Millar ME, Potocek EA, Wing HJ. Investigating the DNA-Binding Site for VirB, a Key Transcriptional Regulator of Shigella Virulence Genes, Using an In Vivo Binding Tool. Genes. 2019; 10(2):149. https://doi.org/10.3390/genes10020149
Chicago/Turabian StyleKarney, Monika M.A., Joy A. McKenna, Natasha Weatherspoon-Griffin, Alexander D. Karabachev, Makensie E. Millar, Eliese A. Potocek, and Helen J. Wing. 2019. "Investigating the DNA-Binding Site for VirB, a Key Transcriptional Regulator of Shigella Virulence Genes, Using an In Vivo Binding Tool" Genes 10, no. 2: 149. https://doi.org/10.3390/genes10020149