Skim-Sequencing Reveals the Likely Origin of the Enigmatic Endangered Sunflower Helianthus schweinitzii

 , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Material, DNA Extraction, and Sequencing
2.2. Assembly of Organelle and Nuclear DNA Regions
2.3. Phylogeny Reconstruction
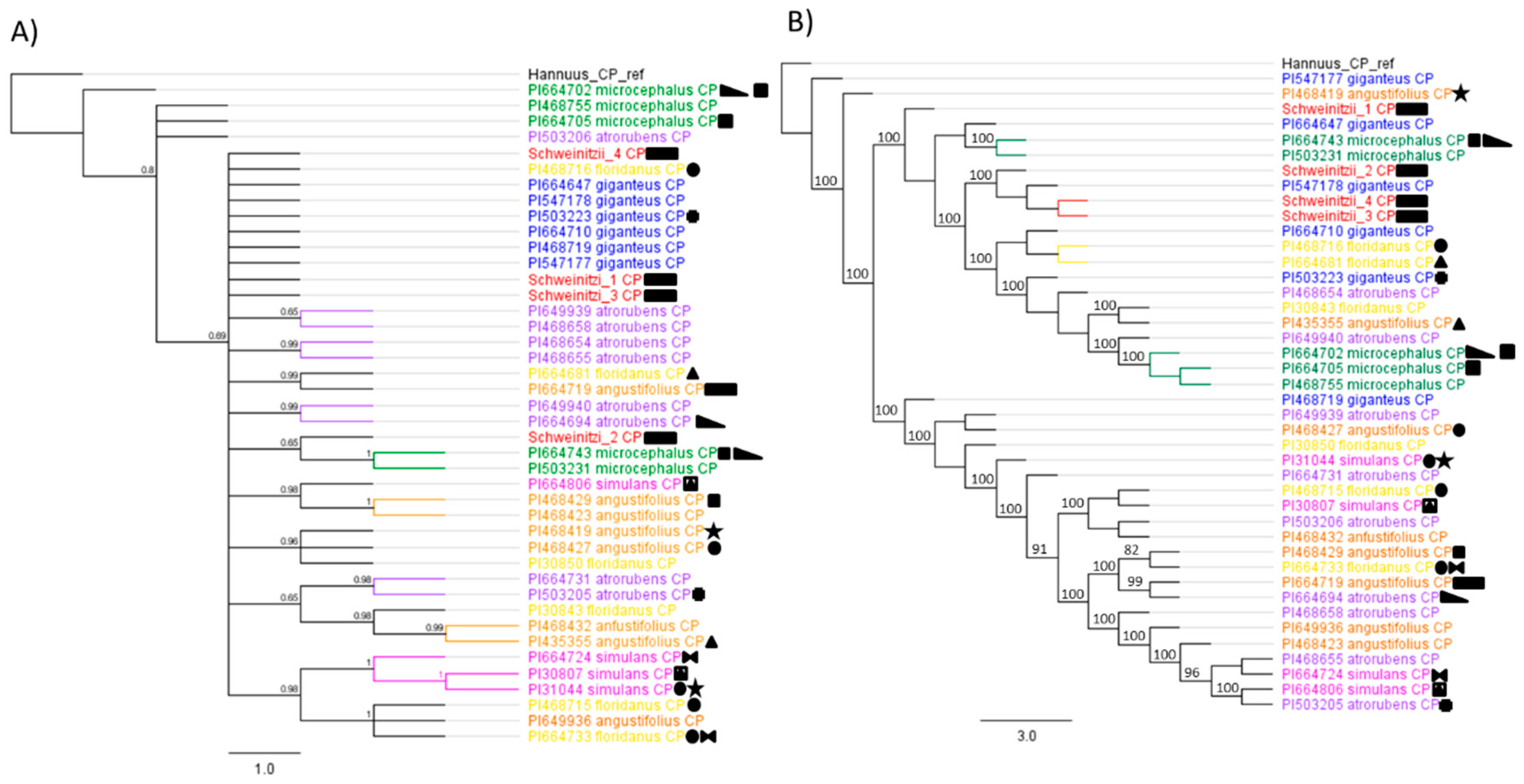
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Hamrick, J.L.; Godt, M.J.W.; Murawski, D.A.; Loveless, M.D. Correlations between species traits and allozyme diversity: Implications for conservation biology. In Genetics and Conservation of Rare Plants; Falk, D.A., Holsinger, K.E., Eds.; Oxford University Press: New York, NY, USA, 1991; pp. 75–86. [Google Scholar]
- Rieseberg, L.H.; Doyle, M.F. Allozyme variation in Helianthus praecox ssp. Hirtus, a rare sunflower from Southern Texas. Aliso 1989, 12, 379–386. [Google Scholar] [CrossRef] [Green Version]
- Shafer, A.B.A.; Wolf, J.B.; Alves, P.C.; Bergström, L.; Bruford, M.W.; Brännström, I.; Colling, G.; Dalén, L.; De Meester, L.; Ekblom, R.; et al. Genomics and the challenging translation into conservation practice. Trends Ecol. Evol. 2015, 30, 78–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crandall, K.A.; Bininda-Emonds, O.R.; Mace, G.M.; Wayne, R.K. Considering evolutionary processes in conservation biology. Trends Ecol. Evol. 2000, 15, 290–295. [Google Scholar] [CrossRef]
- Funk, W.C.; McKay, J.K.; Hohenlohe, P.A.; Allendorf, F.W. Harnessing genomics for delineating conservation units. Trends Ecol. Evol. 2012, 27, 489–496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avise, J.C. A role for molecular genetics in the recognition and conservation of endangered species. Trends Ecol. Evol. 1989, 4, 279–281. [Google Scholar] [CrossRef] [Green Version]
- Schwartz, M.K.; Luikart, G.; Waples, R.S. Genetic monitoring as a promising tool for conservation and management. Trends Ecol. Evol. 2007, 22, 25–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Todesco, M.; Pascual, M.A.; Owens, G.L.; Ostevik, K.L.; Moyers, B.T.; Hübner, S.; Heredia, S.M.; Hahn, M.A.; Caseys, C.; Bock, D.G.; et al. Hybridization and extinction. Evol. Appl. 2016, 9, 892–908. [Google Scholar] [CrossRef]
- Pimm, S.L.; Dollar, L.; Bass, O.L., Jr. The genetic rescue of the Florida panther. Anim. Conserv. 2006, 9, 115–122. [Google Scholar] [CrossRef]
- Chan, W.Y.; Hoffmann, A.A.; van Oppen, M.J. Hybridization as a conservation management tool. Conserv. Lett. 2019, e12652. [Google Scholar] [CrossRef]
- Quinzin, M.C.; Sandoval-Castillo, J.; Miller, J.M.; Beheregaray, L.B.; Russello, M.A.; Hunter, E.A.; Gibbs, J.P.; Tapia, W.; Villalva, F.; Caccone, A. Genetically informed captive breeding of hybrids of an extinct species of Galapagos tortoise. Conserv. Biol. 2019. [Google Scholar] [CrossRef]
- Rieseberg, L.H.; Willis, J.H. Plant speciation. Science 2007, 317, 910–914. [Google Scholar] [CrossRef] [PubMed]
- Rieseberg, L.H. Major ecological transitions in wild sunflowers facilitated by hybridization. Science 2003, 301, 1211–1216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baack, E.J.; Whitney, K.D.; Rieseberg, L.H. Hybridization and genome size evolution: Timing and magnitude of nuclear DNA content increases in Helianthus homoploid hybrid species. New Phytol. 2005, 167, 623–630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buggs, R.J.; Wendel, J.F.; Doyle, J.J.; Soltis, D.E.; Soltis, P.S.; Coate, J.E. The legacy of diploid progenitors in allopolyploid gene expression patterns. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2014, 369. [Google Scholar] [CrossRef] [Green Version]
- Soltis, D.E.; Soltis, P.S. Polyploidy and Genome Evolution; Springer: Berlin, Germany, 2012. [Google Scholar]
- Wagner, C.E.; Keller, I.; Wittwer, S.; Selz, O.M.; Mwaiko, S.; Greuter, L.; Sivasundar, A.; Seehausen, O. Genome-wide RAD sequence data provide unprecedented resolution of species boundaries and relationships in the Lake Victoria cichlid adaptive radiation. Mol. Ecol. 2013, 22, 787–798. [Google Scholar] [CrossRef]
- Kane, N.; Sveinsson, S.; Dempewolf, H.; Yang, J.Y.; Zhang, D.; Engels, J.M.; Cronk, Q. Ultra-barcoding in cacao (Theobroma spp.; Malvaceae) using whole chloroplast genomes and nuclear ribosomal DNA. Am. J. Bot. 2012, 99, 320–329. [Google Scholar] [CrossRef] [Green Version]
- Straub, S.C.; Parks, M.; Weitemier, K.; Fishbein, M.; Cronn, R.C.; Liston, A. Navigating the tip of the genomic iceberg: Next generation sequencing for plant systematics. Am. J. Bot. 2012, 99, 349–364. [Google Scholar] [CrossRef] [Green Version]
- Dodsworth, S. Genome skimming for next-generation biodiversity analysis. Trends Plant Sci. 2015, 20, 525–527. [Google Scholar] [CrossRef]
- Bock, D.G.; Kane, N.C.; Ebert, D.P.; Rieseberg, L.H. Genome skimming reveals the origin of the Jerusalem Artichoke tuber crop species: Neither from Jerusalem nor an artichoke. New Phytol. 2014, 201, 1021–1030. [Google Scholar] [CrossRef]
- Malé, P.J.G.; Bardon, L.; Besnard, G.; Coissac, E.; Delsuc, F.; Engel, J.; Lhuillier, E.; Scotti-Saintagne, C.; Tinaut, A.; Chave, J. Genome skimming by shotgun sequencing helps resolve the phylogeny of a pantropical tree family. Mol. Ecol. Resour. 2014, 14, 966–975. [Google Scholar]
- Mariac, C.; Scarcelli, N.; Pouzadou, J.; Barnaud, A.; Billot, C.; Faye, A.; Kougbeadjo, A.; Maillol, V.; Martin, G.; Sabot, F.; et al. Cost-effective enrichment hybridization capture of chloroplast genomes at deep multiplexing levels for population genetics and phylogeography studies. Mol. Ecol. Resour. 2014, 14, 1103–1113. [Google Scholar] [CrossRef] [PubMed]
- Barker, M.S.; Kane, N.C.; Matvienko, M.; Kozik, A.; Michelmore, R.W.; Knapp, S.J.; Rieseberg, L.H. Multiple paleopoly- ploidizations during the evolution of the Compositae reveal parallel patterns of duplicate gene retention after millions of years. Molec. Biol. Evol. 2008, 25, 2445–2455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Timme, R.E.; Simpson, B.B.; Linder, C.R. High-resolution phylogeny for Helianthus (Asteraceae) using the 18S-26S ribosomal DNA external transcribed spacer. Am. J. Bot. 2007, 94, 1837–1852. [Google Scholar] [CrossRef] [PubMed]
- Schilling, E.E.; Heiser, C.B. Infrageneric classification of Helianthus (Compositae). Taxon 1981, 30, 393–403. [Google Scholar] [CrossRef]
- Baute, G.J.; Owens, G.L.; Bock, D.G.; Rieseberg, L.H. Genome-wide genotyping-by-sequencing data provide a high-resolution view of wild Helianthus diversity, genetic structure, and interspecies gene flow. Am. J. Bot. 2016, 103, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stephens, J.; Rogers, W.; Mason, C.; Donovan, L.; Malmberg, R. Species tree estimation of diploid Helianthus (Asteraceae) using target enrichment. Am. J. Bot. 2015, 102, 910–920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Federal Registry. Helianthus schweinitzii (Schweinitz’s Sunflower) Determined to be Endangered; 56 Fed. Reg. No. 88 (Tuesday, May 7, 1991); National Archives and Records Administration: College Park, MA, USA, 1991; pp. 21087–21091.
- Matthews, J.F.; Howard, J.H. Genetic Variation in the Federally Endangered Schweinitz’s Sunflower, Helianthus schweinitzii T. & G. (Asteraceae). Castanea 1999, 64, 231–242. [Google Scholar]
- Heiser, C.B.; Smith, D.M.; Clevenger, S.B.; Martin, W.C. The American sunflowers (Helianthus). Mem. Torrey Bot. Club 1969, 22, 1–213. [Google Scholar]
- Kane, N.C.; Burke, J.M.; Marek, L.; Seiler, G.; Vear, F.; Baute, G.; Knapp, S.J.; Vincourt, P.; Rieseberg, L.H. Sunflower genetic, genomic and ecological resources. Mol. Ecol. Resour. 2013, 13, 10–20. [Google Scholar] [CrossRef]
- Qiu, F.; Baack, E.J.; Whitney, K.D.; Bock, D.G.; Tetreault, H.M.; Rieseberg, L.H.; Ungerer, M.C. Phylogenetic trends and environmental correlates of nuclear genome size variation in Helianthus sunflowers. New Phytol. 2019, 221, 1609–1618. [Google Scholar] [CrossRef] [Green Version]
- Torrey, J.; Gray, A. A Flora of North America; Wiley and Putnam: New York, NY, USA, 1842; Volume II, Part II. [Google Scholar]
- Grubbs, K.C.; Wynes, A. Reproductive biology of the endangered Schweinitz’s sunflower (Helianthus schweinitzii). Castanea 2015, 80, 20–28. [Google Scholar] [CrossRef]
- Rogers, C.E.; Thompson, T.E.; Seiler, G.J. Sunflower Species of the United States; National Sunflower Association: Bismarck, ND, USA, 1982; pp. 1–75. [Google Scholar]
- Heiser, C.B.; Martin, W.C.; Smith, D.M. Species crosses in Helianthus: I. Diploid species. Brittonia 1962, 14, 137–147. [Google Scholar] [CrossRef]
- Heiser, C.B.; Smith, D.M. Species crosses in Helianthus: II. Polyploid species. Rhodora 1964, 66, 344–358. [Google Scholar]
- Chandler, J.M.; Jan, C.C.; Beard, B.H. Chromosomal differentiation among the annual Helianthus species. Syst. Bot. 1986, 354–371. [Google Scholar] [CrossRef]
- Matthews, J.F.; Barden, L.S.; Matthews, C.R. Corrections of the chromosome number, distribution, and misidentifications of the federally endangered sunflower, Helianthus schweinitzii T. & G. J. Torrey Bot. Soc. 1997, 124, 198–209. [Google Scholar]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014. [Google Scholar] [CrossRef] [Green Version]
- Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv 2013, arXiv:1303.3997. [Google Scholar]
- Zerbino, D.R.; Birney, E. Velvet: Algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008, 18, 821–829. [Google Scholar] [CrossRef] [Green Version]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef]
- Katoh, K.; Toh, H. Recent developments in the MAFFT multiple sequence alignment program. Briefings in Bioinformatics 2008, 9, 286–298. [Google Scholar] [CrossRef] [Green Version]
- Guindon, S.; Gascuel, O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst. Biol. 2003, 52, 696–704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ronquist, F.; Huelsenbeck, J.P. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 2003, 19, 1572–1574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamura, K.; Nei, M. Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol. Biol. Evol. 1993, 10, 512–526. [Google Scholar] [PubMed]
- Timme, R.E.; Kuehl, J.V.; Boore, J.L.; Jansen, R.K. A comparative analysis of the Lactuca and Helianthus (Asteraceae) plastid genomes: Identification of divergent regions and categorization of shared repeats. Am. J. Bot. 2007, 94, 302–312. [Google Scholar] [CrossRef]
- Guggisberg, A.; Bretagnolle, F.; Mansion, G. Allopolyploid origin of the Mediterranean endemic, Centaurium bianoris (Gentianaceae), inferred by molecular markers. Syst. Bot. 2006, 31, 368–379. [Google Scholar] [CrossRef]
- Slotte, T.; Ceplitis, A.; Neuffer, B.; Hurka, H.; Lascoux, M. Intrageneric phylogeny of Capsella (Brassicaceae) and the origin of the tetraploid C. bursa-pastoris based on chloroplast and nuclear DNA sequences. Am. J. Bot. 2006, 93, 1714–1724. [Google Scholar] [CrossRef]
- Lee-Yaw, J.A.; Grassa, C.J.; Joly, S.; Andrew, R.L.; Rieseberg, L.H. An evaluation of alternative explanations for widespread cytonuclear discordance in annual sunflowers (Helianthus). New Phytol. 2019, 221, 515–526. [Google Scholar] [CrossRef] [Green Version]
- Maddison, W.P.; Knowles, L.L. Inferring phylogeny despite incomplete lineage sorting. Syst. Biol. 2006, 55, 21–30. [Google Scholar] [CrossRef]
- Pollard, D.A.; Iyer, V.N.; Moses, A.M.; Eisen, M.B. Widespread discordance of gene trees with species tree in Drosophila: Evidence for incomplete lineage sorting. PLoS Genet. 2006, 2, e173. [Google Scholar] [CrossRef] [Green Version]
- Schilling, E.E. Phylogenetic analysis of Helianthus (Asteraceae) based on chloroplast DNA restriction site data. Theor. Appl. Genet. 1997, 94, 925–933. [Google Scholar] [CrossRef]
- Rieseberg, L.H.; Soltis, D.E. Phylogenetic consequences of cytoplasmic gene flow in plants. Evol. Trends Plants 1991, 5, 65–84. [Google Scholar]
- Soltis, D.E.; Soltis, P.S. Polyploidy: Recurrent formation and genome evolution. Trends Ecol. Evol. 1999, 14, 348–352. [Google Scholar] [CrossRef]
- El Baidouri, M.; Murat, F.; Veyssiere, M.; Molinier, M.; Flores, R.; Burlot, L.; Alaux, M.; Quesneville, H.; Pont, C.; Salse, J. Reconciling the evolutionary origin of bread wheat (Triticum aestivum). New Phytol. 2017, 213, 1477–1486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spring, O.; Schilling, E.E. The sesquiterpene lactone chemistry of Helianthus sect. Atrorubentes (Asteraceae: Heliantheae). Biochem. Syst. Ecol. 1991, 19, 59–79. [Google Scholar] [CrossRef]
- Rieseberg, L.H.; Ellstrand, N.C. What can morphological and molecular markers tell us about plant hybridization? Crit. Rev. Plant Sci. 1993, 12, 213–241. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Helianthus atrorubens | Helianthus floridanus | Helianthus giganteus | Helianthus microcephalus | Helianthus simulans | Helianthus schweinitzii | |
|---|---|---|---|---|---|---|
| H. angustifolius | Y | Y | N | N | Y | Y |
| H. atrorubens | N | Y | Y | Y | N | |
| H. floridanus | N | N | Y | N | ||
| H. giganteus | Y | N | Y | |||
| H. microcephalus | N | NA | ||||
| H. simulans | NA |
| Accession | Species | Range | Latitude | Longitude |
|---|---|---|---|---|
| PI468419 | H. angustifolius | East Central USA | 29°39′0″N | 82°19′0″W |
| PI435355 | H. angustifolius | East Central USA | 32°3′0″N | 84°11′W |
| PI468423 | H. angustifolius | East Central USA | 33°32′N | 92°28′0″W |
| PI468427 | H. angustifolius | East Central USA | 30°49′0″N | 82°0′0″W |
| PI468429 | H. angustifolius | East Central USA | 34°18′0″N | 79°2′W |
| PI468432 | H. angustifolius | East Central USA | 34°19′0″N | 78°30′0″W |
| PI649936 | H. angustifolius | East Central USA | 35°23′57″N | 86°1′0″W |
| PI664719 | H. angustifolius | East Central USA | 35°18′53″N | 80°2′49″W |
| PI503206 | H. atrorubens | East Central USA | 37°0′0″N | 77°0′0″W |
| PI649939 | H. atrorubens | East Central USA | 36°36′30″N | 88°41′30″W |
| PI649940 | H. atrorubens | East Central USA | 33°53′26″N | 86°49′33″W |
| PI664694 | H. atrorubens | East Central USA | 34°39′37″N | 83°20′53″W |
| PI664731 | H. atrorubens | East Central USA | 33°11′40″N | 79°31′32″W |
| PI503205 | H. atrorubens | East Central USA | 36°0′0″N | 77°0′0″W |
| PI468654 | H. atrorubens | East Central USA | 33°1′0″N | 84°42′0″W |
| PI468655 | H. atrorubens | East Central USA | 34°14′N | 84°29′W |
| PI468658 | H. atrorubens | East Central USA | 33°49′0″N | 81°6′0″W |
| PI664733 | H. floridanus | South East USA | 31°32′51″N | 81°32′24″W |
| PI30843 | H. floridanus | South East USA | 29°42′53″N | 85°1′31″W |
| PI30850 | H. floridanus | South East USA | 28°40′30″N | 80°58′34″W |
| PI468715 | H. floridanus | South East USA | 30°33′0″N | 81°49′0″W |
| PI468716 | H. floridanus | South East USA | 30°49′0″N | 82°0′0″W |
| PI664681 | H. floridanus | South East USA | 31°18′17″N | 83°48′40″W |
| PI547177 | H. giganteus | East Central USA | 46°37′00″N | 90°46′00″W |
| PI664647 | H. giganteus | East Central USA | 41°35′27″N | 83°45′43″W |
| PI664710 | H. giganteus | East Central USA | 35°48′42″N | 82°11′50″W |
| PI468719 | H. giganteus | East Central USA | 36°18′00″N | 78°35′00″W |
| PI547178 | H. giganteus | East Central USA | 45°15′00″N | 88°36′00″W |
| PI503223 | H. giganteus | East Central USA | 36°00′00″N | 77°00′00″W |
| PI664743 | H. microcephalus | East Central USA | 34°15′45″N | 82°39′46″W |
| PI468756 | H. microcephalus | East Central USA | 36°7′0″N | 79°25′0″W |
| PI503231 | H. microcephalus | East Central USA | 37°0′0″N | 80°0′0″W |
| PI664702 | H. microcephalus | East Central USA | 34°56′51″N | 83°5′21″W |
| PI664705 | H. microcephalus | East Central USA | 35°10′56″N | 82°22′15″W |
| PI31044 | H. simulans | South East USA | 29°58′50″N | 82°14′12″W |
| PI30807 | H. simulans | South East USA | 30°28′58″N | 90°55′11″W |
| PI664724 | H. simulans | South East USA | 32°7′28″N | 81°37′25″W |
| PI664806 | H. simulans | South East USA | 30°27′8″N | 90°54′52″W |
| NA | H. schweinitzii_01 | Piedmont plateau in North and South Carolina | 34°54′7″N | 81°1′18″W |
| NA | H. schweinitzii_02 | Piedmont plateau in North and South Carolina | 34°54′7″N | 81°1′19″W |
| NA | H. schweinitzii_03 | Piedmont plateau in North and South Carolina | 34°56′29″N | 81°0′28″W |
| NA | H. schweinitzii_04 | Piedmont plateau in North and South Carolina | 34°56′29″N | 81°0′28″W |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Anderson, J.; Kantar, M.; Bock, D.; Grubbs, K.C.; Schilling, E.; Rieseberg, L. Skim-Sequencing Reveals the Likely Origin of the Enigmatic Endangered Sunflower Helianthus schweinitzii. Genes 2019, 10, 1040. https://doi.org/10.3390/genes10121040
Anderson J, Kantar M, Bock D, Grubbs KC, Schilling E, Rieseberg L. Skim-Sequencing Reveals the Likely Origin of the Enigmatic Endangered Sunflower Helianthus schweinitzii. Genes. 2019; 10(12):1040. https://doi.org/10.3390/genes10121040
Chicago/Turabian StyleAnderson, Justin, Michael Kantar, Dan Bock, Kunsiri Chaw Grubbs, Edward Schilling, and Loren Rieseberg. 2019. "Skim-Sequencing Reveals the Likely Origin of the Enigmatic Endangered Sunflower Helianthus schweinitzii" Genes 10, no. 12: 1040. https://doi.org/10.3390/genes10121040