Nanopore Targeted Sequencing for Rapid Gene Mutations Detection in Acute Myeloid Leukemia


 ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patients
2.2. Acute Myeloid Leukemia Panel Design and Testing
2.3. Multiplex Long-Polymerase Chain Reaction
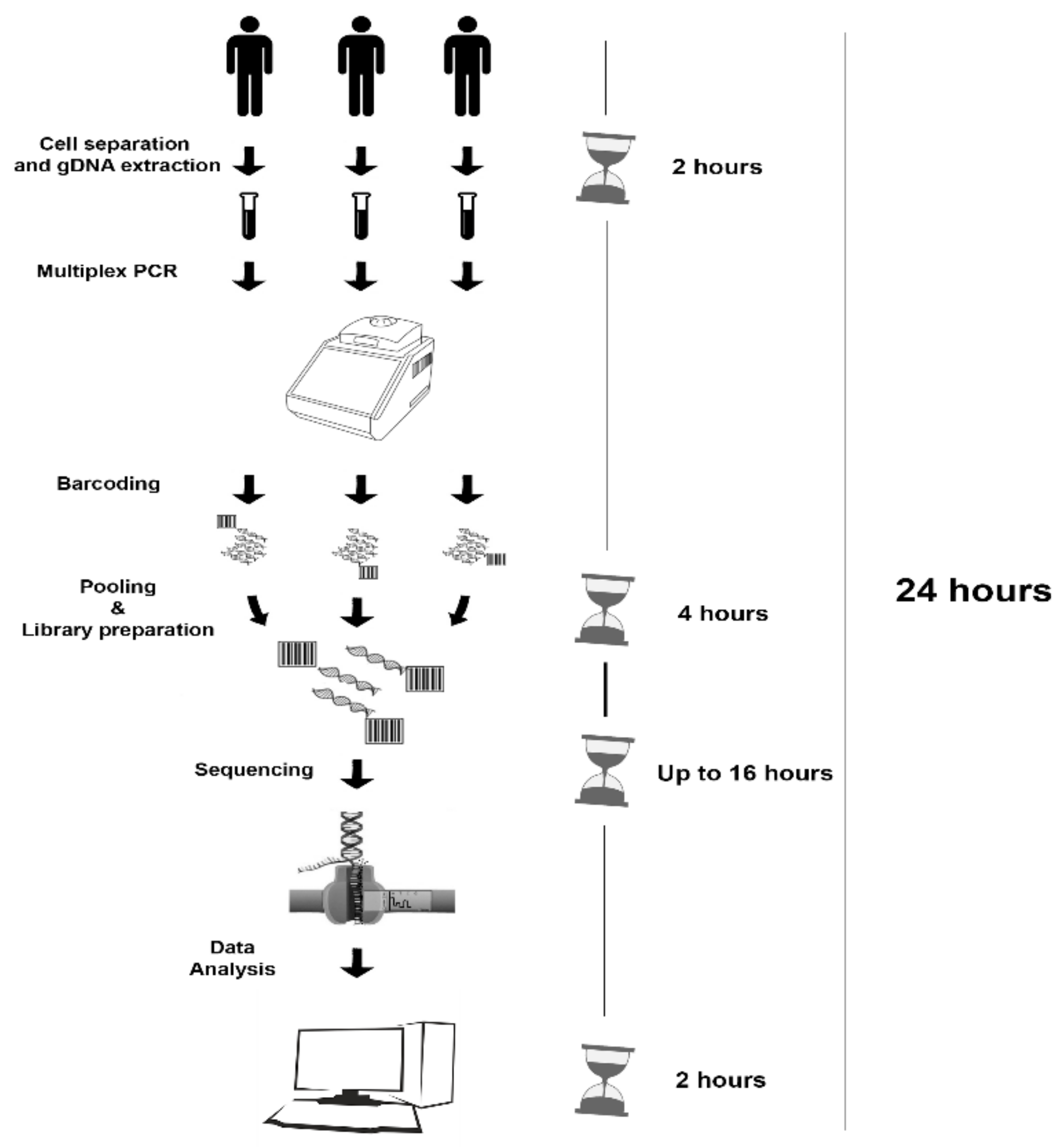
2.4. MinION Sequencing (MS)
2.5. MinION Sequencing Data Analysis
2.6. S5 Sequencing (S5S)
2.7. Sanger Sequencing (SS)
2.8. Data Availability
3. Results
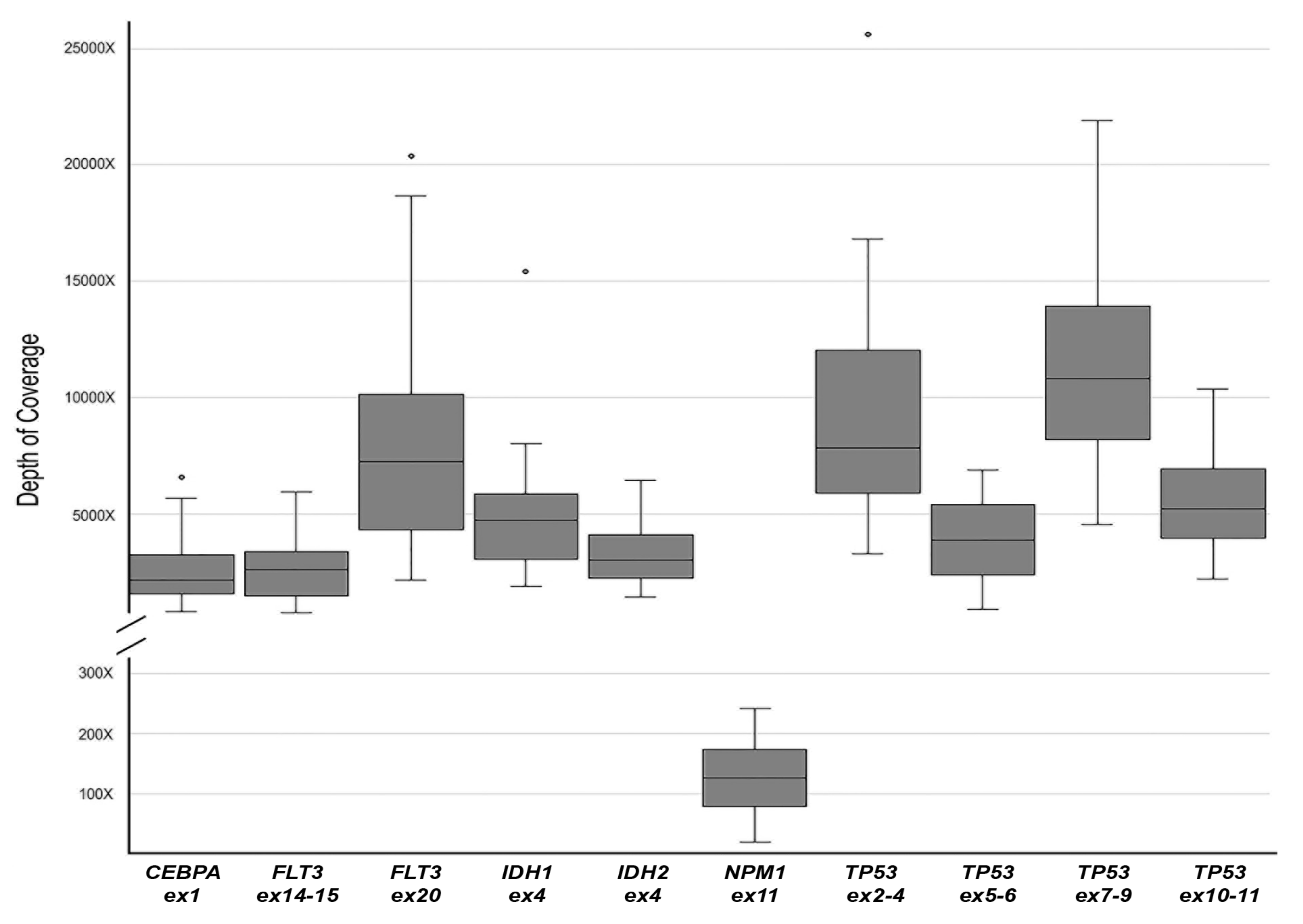
3.1. MinION Sequencing Performance Evaluation
3.2. Variant Calling, Filtering and Annotation
3.3. Identification of the Acute Myeloid Leukemia Hotspot Mutations
3.4. FLT3 Internal Tandem Duplications (ITD) Data Analysis
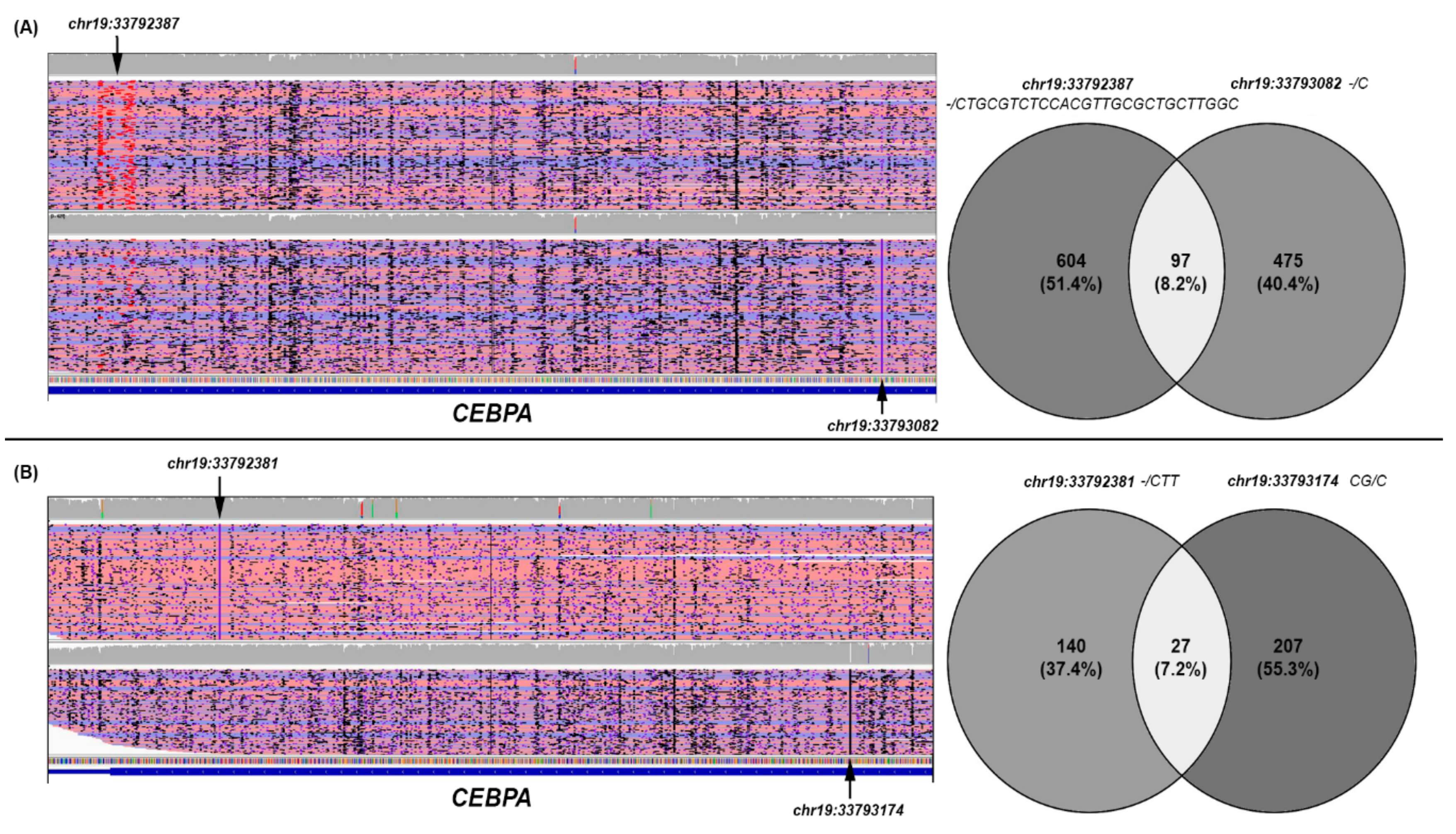
3.5. CEBPA Data Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016, 127, 2391–2405. [Google Scholar] [CrossRef] [PubMed]
- Döhner, H.; Estey, E.; Grimwade, D.; Amadori, S.; Appelbaum, F.R.; Büchner, T.; Dombret, H.; Ebert, B.L.; Fenaux, P.; Larson, R.A.; et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood 2017, 129, 424–447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Döhner, H.; Estey, E.H.; Amadori, S.; Appelbaum, F.R.; Büchner, T.; Burnett, A.K.; Dombret, H.; Fenaux, P.; Grimwade, D.; Larson, R.A.; et al. Diagnosis and management of acute myeloid leukemia in adults: Recommendations from an international expert panel, on behalf of the European LeukemiaNet. Blood 2010, 115, 453–474. [Google Scholar] [CrossRef] [PubMed]
- Stone, R.M.; Mandrekar, S.J.; Sanford, B.L.; Laumann, K.; Geyer, S.; Bloomfield, C.D.; Thiede, C.; Prior, T.W.; Döhner, K.; Marcucci, G.; et al. Midostaurin plus chemotherapy for acute myeloid leukemia with a FLT3 Mutation. N. Engl. J. Med. 2017, 377, 454–464. [Google Scholar] [CrossRef]
- DiNardo, C.D.; Stein, E.M.; de Botton, S.; Roboz, G.J.; Altman, J.K.; Mims, A.S.; Swords, R.; Collins, R.H.; Mannis, G.N.; Pollyea, D.A.; et al. Durable remissions with Ivosidenib in IDH1-Mutated relapsed or refractory AML. N. Engl. J. Med. 2018, 378, 2386–2398. [Google Scholar] [CrossRef]
- Stein, E.M.; DiNardo, C.D.; Pollyea, D.A.; Fathi, A.T.; Roboz, G.J.; Altman, J.K.; Stone, R.M.; DeAngelo, D.J.; Levine, R.L.; Flinn, I.W.; et al. Enasidenib in mutant IDH2 relapsed or refractory acute myeloid leukemia. Blood 2017, 130, 722–731. [Google Scholar] [CrossRef]
- Lu, H.; Giordano, F.; Ning, Z. Oxford nanopore MinION sequencing and genome assembly. Genom. Proteom. Bioinform. 2016, 14, 265–279. [Google Scholar] [CrossRef] [Green Version]
- Minervini, C.F.; Cumbo, C.; Orsini, P.; Brunetti, C.; Anelli, L.; Zagaria, A.; Minervini, A.; Casieri, P.; Coccaro, N.; Tota, G.; et al. TP53 gene mutation analysis in chronic lymphocytic leukemia by nanopore MinION sequencing. Diagn. Pathol. 2016, 11, 96. [Google Scholar] [CrossRef] [Green Version]
- Minervini, C.F.; Cumbo, C.; Orsini, P.; Anelli, L.; Zagaria, A.; Impera, L.; Coccaro, N.; Brunetti, C.; Minervini, A.; Casieri, P.; et al. Mutational analysis in BCR-ABL1 positive leukemia by deep sequencing based on nanopore MinION technology. Exp. Mol. Pathol. 2017, 103, 33–37. [Google Scholar] [CrossRef]
- Cumbo, C.; Impera, L.; Minervini, C.F.; Orsini, P.; Anelli, L.; Zagaria, A.; Coccaro, N.; Tota, G.; Minervini, A.; Casieri, P.; et al. Genomic BCR-ABL1 breakpoint characterization by a multi-strategy approach for “personalized monitoring” of residual disease in chronic myeloid leukemia patients. Oncotarget 2018, 9, 10978–10986. [Google Scholar] [CrossRef] [Green Version]
- Orsini, P.; Minervini, C.F.; Cumbo, C.; Anelli, L.; Zagaria, A.; Minervini, A.; Coccaro, N.; Tota, G.; Casieri, P.; Impera, L.; et al. Design and MinION testing of a nanopore targeted gene sequencing panel for chronic lymphocytic leukemia. Sci. Rep. 2018, 8, 11798. [Google Scholar] [CrossRef]
- Rücker, F.G.; Schlenk, R.F.; Bullinger, L.; Kayser, S.; Teleanu, V.; Kett, H.; Habdank, M.; Kugler, C.-M.; Holzmann, K.; Gaidzik, V.I.; et al. TP53 alterations in acute myeloid leukemia with complex karyotype correlate with specific copy number alterations, monosomal karyotype, and dismal outcome. Blood 2011, 118. [Google Scholar] [CrossRef]
- Albano, F.; Anelli, L.; Zagaria, A.; Coccaro, N.; D’Addabbo, P.; Liso, V.; Rocchi, M.; Specchia, G. Genomic segmental duplications on the basis of the t(9;22) rearrangement in chronic myeloid leukemia. Oncogene 2010, 29, 2509–2516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albano, F.; Anelli, L.; Zagaria, A.; Coccaro, N.; Minervini, A.; Rossi, A.R.; Specchia, G. Decreased TET2 gene expression during chronic myeloid leukemia progression. Leuk. Res. 2011, 35, e220–e222. [Google Scholar] [CrossRef] [PubMed]
- Storlazzi, C.T.; Albano, F.; Lo Cunsolo, C.; Doglioni, C.; Guastadisegni, M.C.; Impera, L.; Lonoce, A.; Funes, S.; Macrì, E.; Iuzzolino, P.; et al. Upregulation of the SOX5 by promoter swapping with the P2RY8 gene in primary splenic follicular lymphoma. Leukemia 2007, 21, 2221–2225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Storlazzi, C.T.; Albano, F.; Locunsolo, C.; Lonoce, A.; Funes, S.; Guastadisegni, M.C.; Cimarosto, L.; Impera, L.; D’Addabbo, P.; Panagopoulos, I.; et al. t(3;12)(q26;q14) in polycythemia vera is associated with upregulation of the HMGA2 gene. Leukemia 2006, 20, 2190–2192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Durbin, R. Fast and accurate long-read alignment with Burrows–Wheeler transform. Bioinformatics 2010, 26, 589–595. [Google Scholar] [CrossRef] [Green Version]
- Robinson, J.T.; Thorvaldsdóttir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative genomics viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef] [Green Version]
- Sedlazeck, F.J.; Rescheneder, P.; Smolka, M.; Fang, H.; Nattestad, M.; von Haeseler, A.; Schatz, M.C. Accurate detection of complex structural variations using single-molecule sequencing. Nat. Methods 2018, 15, 461–468. [Google Scholar] [CrossRef] [Green Version]
- Huang, X.; Madan, A. CAP3: A DNA sequence assembly program. Genome Res. 1999, 9, 868–877. [Google Scholar] [CrossRef] [Green Version]
- Coccaro, N.; Zagaria, A.; Orsini, P.; Anelli, L.; Tota, G.; Casieri, P.; Impera, L.; Minervini, A.; Minervini, C.F.; Cumbo, C.; et al. RARA and RARG gene downregulation associated with EZH2 mutation in acute promyelocytic-like morphology leukemia. Hum. Pathol. 2018, 80, 82–86. [Google Scholar] [CrossRef] [PubMed]
- Kiyoi, H.; Naoe, T.; Nakano, Y.; Yokota, S.; Minami, S.; Miyawaki, S.; Asou, N.; Kuriyama, K.; Jinnai, I.; Shimazaki, C.; et al. Prognostic implication of FLT3 and N-RAS gene mutations in acute myeloid leukemia. Blood 1999, 93, 3074–3080. [Google Scholar] [PubMed]
- Magi, A.; Semeraro, R.; Mingrino, A.; Giusti, B.; D’Aurizio, R. Nanopore sequencing data analysis: State of the art, applications and challenges. Brief. Bioinform. 2017, 19, 1256–1272. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, C.R.; Wang, H.; Dunbar, W.B. Error analysis of idealized nanopore sequencing. Electrophoresis 2013, 34, 2137–2144. [Google Scholar] [CrossRef] [Green Version]
- Jain, M.; Fiddes, I.T.; Miga, K.H.; Olsen, H.E.; Paten, B.; Akeson, M. Improved data analysis for the MinION nanopore sequencer. Nat. Methods 2015, 12, 351–356. [Google Scholar] [CrossRef] [Green Version]
- Eckert, S.E.; Chan, J.Z.M.; Houniet, D.; Breuer, J.; Speight, G. Enrichment by hybridisation of long DNA fragments for Nanopore sequencing. Microb. Genom. 2016, 2, e000087. [Google Scholar] [CrossRef] [Green Version]
- Gilpatrick, T.; Lee, I.; Graham, J.E.; Raimondeau, E.; Bowen, R.; Heron, A.; Sedlazeck, F.J.; Timp, W. Targeted nanopore sequencing with Cas9 for studies of methylation, structural variants, and mutations. bioRxiv 2019, 604173. [Google Scholar] [CrossRef] [Green Version]
- Schlenk, R.F.; Kayser, S.; Bullinger, L.; Kobbe, G.; Casper, J.; Ringhoffer, M.; Held, G.; Brossart, P.; Lübbert, M.; Salih, H.R.; et al. Differential impact of allelic ratio and insertion site in FLT3-ITD-positive AML with respect to allogeneic transplantation. Blood 2014, 124, 3441–3449. [Google Scholar] [CrossRef] [Green Version]
- Arreba-Tutusaus, P.; Mack, T.S.; Bullinger, L.; Schnöder, T.M.; Polanetzki, A.; Weinert, S.; Ballaschk, A.; Wang, Z.; Deshpande, A.J.; Armstrong, S.A.; et al. Impact of FLT3-ITD location on sensitivity to TKI-therapy in vitro and in vivo. Leukemia 2016, 30, 1220–1225. [Google Scholar] [CrossRef]
- Liu, S.-B.; Dong, H.-J.; Bao, X.-B.; Qiu, Q.-C.; Li, H.-Z.; Shen, H.-J.; Ding, Z.-X.; Wang, C.; Chu, X.-L.; Yu, J.-Q.; et al. Impact of FLT3 -ITD length on prognosis of acute myeloid leukemia. Haematologica 2019, 104, e9–e12. [Google Scholar] [CrossRef] [Green Version]
- Schranz, K.; Hubmann, M.; Harin, E.; Vosberg, S.; Herold, T.; Metzeler, K.H.; Rothenberg-Thurley, M.; Janke, H.; Bräundl, K.; Ksienzyk, B.; et al. Clonal heterogeneity of FLT3-ITD detected by high-throughput amplicon sequencing correlates with adverse prognosis in acute myeloid leukemia. Oncotarget 2018, 9, 30128–30145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krishnakumar, R.; Sinha, A.; Bird, S.W.; Jayamohan, H.; Edwards, H.S.; Schoeniger, J.S.; Patel, K.D.; Branda, S.S.; Bartsch, M.S. Systematic and stochastic influences on the performance of the MinION nanopore sequencer across a range of nucleotide bias. Sci. Rep. 2018, 8, 3159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Green, C.L.; Koo, K.K.; Hills, R.K.; Burnett, A.K.; Linch, D.C.; Gale, R.E. Prognostic significance of CEBPA mutations in a large cohort of younger adult patients with acute myeloid leukemia: Impact of double CEBPA mutations and the interaction with FLT3 and NPM1 mutations. J. Clin. Oncol. 2010, 28, 2739–2747. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M.; Sukhai, M.A.; Zhang, T.; Dolatshahi, R.; Harbi, D.; Garg, S.; Misyura, M.; Pugh, T.; Stockley, T.L.; Kamel-Reid, S. Integration of technical, bioinformatic, and variant assessment approaches in the validation of a targeted next-generation sequencing panel for myeloid malignancies. Arch. Pathol. Lab. Med. 2017, 141, 759–775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, B.; Hu, Y.; Ng, C.; Ban, K.H.K.; Tan, T.W.; Huan, P.T.; Lee, P.-L.; Chiu, L.; Seah, E.; Ng, C.H.; et al. Coverage analysis in a targeted amplicon-based next-generation sequencing panel for myeloid neoplasms. J. Clin. Pathol. 2016, 69, 801–804. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, A.; Suzuki, M.; Mizushima-Sugano, J.; Frith, M.C.; Makałowski, W.; Kohno, T.; Sugano, S.; Tsuchihara, K.; Suzuki, Y. Sequencing and phasing cancer mutations in lung cancers using a long-read portable sequencer. DNA Res. 2017, 24, 585–596. [Google Scholar] [CrossRef]
- Cretu Stancu, M.; van Roosmalen, M.J.; Renkens, I.; Nieboer, M.M.; Middelkamp, S.; de Ligt, J.; Pregno, G.; Giachino, D.; Mandrile, G.; Espejo Valle-Inclan, J.; et al. Mapping and phasing of structural variation in patient genomes using nanopore sequencing. Nat. Commun. 2017, 8, 1326. [Google Scholar] [CrossRef] [Green Version]
- Alonso, C.M.; Llop, M.; Sargas, C.; Pedrola, L.; Panadero, J.; Hervás, D.; Cervera, J.; Such, E.; Ibáñez, M.; Ayala, R.; et al. Clinical utility of a next-generation sequencing panel for acute myeloid leukemia diagnostics. J. Mol. Diagn. 2019, 21, 228–240. [Google Scholar] [CrossRef] [Green Version]
- Jennings, L.J.; Arcila, M.E.; Corless, C.; Kamel-Reid, S.; Lubin, I.M.; Pfeifer, J.; Temple-Smolkin, R.L.; Voelkerding, K.V.; Nikiforova, M.N. Guidelines for validation of next-generation sequencing-based oncology panels: A joint consensus recommendation of the association for molecular pathology and college of American pathologists. J. Mol. Diagn. 2017, 19, 341–365. [Google Scholar] [CrossRef] [Green Version]
- Roy, S.; Coldren, C.; Karunamurthy, A.; Kip, N.S.; Klee, E.W.; Lincoln, S.E.; Leon, A.; Pullambhatla, M.; Temple-Smolkin, R.L.; Voelkerding, K.V.; et al. Standards and guidelines for validating next-generation sequencing bioinformatics pipelines. J. Mol. Diagn. 2018, 20, 4–27. [Google Scholar] [CrossRef] [Green Version]
- Tyler, A.D.; Mataseje, L.; Urfano, C.J.; Schmidt, L.; Antonation, K.S.; Mulvey, M.R.; Corbett, C.R. Evaluation of Oxford Nanopore’s MinION sequencing device for microbial whole genome sequencing applications. Sci. Rep. 2018, 8, 10931. [Google Scholar] [CrossRef] [PubMed]
- Patel, K.P.; Ruiz-Cordero, R.; Chen, W.; Routbort, M.J.; Floyd, K.; Rodriguez, S.; Galbincea, J.; Barkoh, B.A.; Hatfield, D.; Khogeer, H.; et al. Ultra-rapid reporting of GENomic targets (URGENTseq): Clinical next-generation sequencing results within 48 hours of sample collection. J. Mol. Diagn. 2019, 21, 89–98. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Pool | Target | Genomic Region | Length (bp) | Primers | Identical Bases Per 100 Aligned Bases | Inserted Bases Per 100 Aligned Bases | Deleted Bases Per 100 Aligned Bases | Substitutions Per 100 Aligned Bases |
|---|---|---|---|---|---|---|---|---|
| 1 | CEBPA exon 1 | chr19:33792118-33793481 | 1363 | 5′-GGAGGCACCGGAATCTCCTA-3′ 5′-GGCCTGCCGGGTATAAAAGCTG-3′ | 86% | 2.9% | 5.6% | 5.4% |
| TP53 exons 7–9 | chr17:7576463-7577667 | 1204 | 5′-CAGGCTAGGCTAAGCTATGATGTTCCTTAGA-3′ 5′-CTTGCCACAGGTCTCCCCAAGG-3′ | 87.8% | 2.5% | 5.6% | 4% | |
| TP53 exons 10–11 | chr17:7572810-7574095 | 1285 | 5′-GTGCTTCTGACGCACACCTATTG-3′ 5′-AACCATCTTTTAACTCAGGTACTGTGT-3′ | 87.5% | 2.5% | 5.9% | 4.1% | |
| 2 | NPM1 exon 11 | chr5:170837410-170837635 | 225 | 5′-GTTAACTCTCTGGTGGTAGAATGAAAAATAGA-3′ 5′-GATATCAACTGTTACAGAAATGAAATAAGACG-3′ | 89.3% | 2.4% | 5.4% | 2.9% |
| FLT3 exons 14–15 | chr13:28607916-28608407 | 491 | 5′-GGCAAACAGTAACCATTAAAAGGATGG-3′ 5′-TTCCTCTATCTGCAGAACTGCCTA-3′ | 88.5% | 3.3% | 4.6% | 3.6% | |
| FLT3 exon 20 | chr13:28592521-28592740 | 219 | 5′-CACAGTGAGTGCAGTTGTTTACCA-3′ 5′-GTCATTCTTGACAGTGTGTTCACAG-3′ | 90.2% | 2.7% | 3.4% | 3.7% | |
| IDH2 exon 4 | chr15:90631713-90632029 | 316 | 5′-CACAAAGTCTGTGGCCTTGTACT-3′ 5′-GTTGAAAGATGGCGGCTGCA-3′ | 88.8% | 2.3% | 5.4% | 3.6% | |
| 3 | TP53 exons 2–4 | chr17:7579204-7579986 | 782 | 5′-GAAGCCAAAGGGTGAAGAGGAATCCC-3′ 5′-AGGGTTGGAAGTGTCTCATGCTGGA-3′ | 86.4% | 2.2% | 7.3% | 4% |
| TP53 exons 5–6 | chr17:7578016-7578702 | 686 | 5′-TTCAACTGTGCAATAGTTAAACCCAT-3′ 5′-CTGAGGTGTAGACGCCAACTCTC-3′ | 87.9% | 2.6% | 5.2% | 4.2% | |
| IDH1 exon 4 | chr2:209113021-209113452 | 431 | 5′-ATACAAGTTGGAAATTTCTGGGCCAT-3′ 5′-CACTGCAGTTGTAGGTTATAACTATCCA-3′ | 88.9% | 2.3% | 4.8% | 3.7% | |
| Mean | 88.1% | 2.6% | 5.3% | 3.9% | ||||
| Case | Gene | Locus | Genotype | Coding Change | Amino Acid Change | Type | Function | Length | Exon | MinION (VAF %) | S5 (VAF %) | Sanger |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| AML#1 | IDH1 | chr2:209113113 | G/A | c.394C | p.R132 | SNV | missense | 1 | 4 | YES (48.9) | YES (47.1) | N/E |
| AML#2 | FLT3 | chr13:28608329 | A/C | c.1727T > G | p.L576R | SNV | missense | 1 | 14 | YES (14.4) | YES (8.3) | N/E |
| AML#3 | NPM1 | chr5:170837547 | -/TCTG | c.859_860insTCTG | p.W288fs*12 | INDEL | Frameshift Insertion | 4 | 11 | YES (35.3) | YES (54.5) | N/E |
| AML#4 | IDH1 | chr2:209113113 | G/A | c.394C | p.R132 | SNV | missense | 1 | 4 | YES (16.9) | YES (14.4) | N/E |
| AML#5 | NPM1 | chr5:170837547 | -/TCTG | c.859_860insTCTG | p.W288fs*12 | INDEL | Frameshift Insertion | 4 | 11 | YES (56.5) | YES (45.8) | N/E |
| IDH2 | chr15:90631934 | C/T | c.419G | p.R140 | SNV | missense | 1 | 4 | YES (32.4) | YES (51.3) | N/E | |
| AML#7 | NPM1 | chr5:170837547 | -/TCTG | c.859_860insTCTG | p.W288fs*12 | INDEL | Frameshift Insertion | 4 | 11 | NO | YES (58.5) | YES |
| IDH2 | chr15:90631934 | C/A | c.419G | p.R140 | SNV | missense | 1 | 4 | YES (38.6) | YES (53.5) | N/E | |
| CEBPA | chr19:33792277 | G/CT | c.1044delinsAG | p.S348fs | INDEL | Frameshift Insertion | 2 | 1 | YES (15.4) | NO | NO | |
| AML#8 | NPM1 | chr5:170837547 | -/TCTG | c.859_860insTCTG | p.W288fs*12 | INDEL | Frameshift Insertion | 4 | 11 | YES (33.3) | YES (46.1) | N/E |
| CEBPA | chr19:33793152 | -/G | c.168dupC | p.E57fs | INDEL | Frameshift Insertion | 1 | 1 | YES (44.3) | NO | YES | |
| AML#9 | NPM1 | chr5:170837547 | -/TCTG | c.859_860insTCTG | p.W288fs*12 | INDEL | Frameshift Insertion | 4 | 11 | YES (31.0) | YES (55.9) | N/E |
| CEBPA | chr19:33792731 | -/GCGGGT | c.589_590insACCCGC | p.H195_P196dup | INDEL | Nonframe shift Insertion | 6 | 1 | NO | YES (18.1) | NO | |
| AML#10 | IDH2 | chr15:90631934 | C/T | c.419G | p.R140 | SNV | missense | 1 | 4 | YES (16.1) | YES (12.1) | N/E |
| AML#13 | CEBPA | chr19:33792387 | -/CTGCGTC TCCACGTTGCGCTGCTTGGC | c.933_934insGCCAAGCAGCGCAACGTGGAGACGCAG | p.A303_Q311dup | INDEL | Nonframe shift Insertion | 27 | 1 | YES (26.2) | YES (38.9) | N/E |
| CEBPA | chr19:33793082 | -/C | c.238dupG | p.D80fs | INDEL | Frame shift Insertion | 1 | 1 | YES (30.4) | NO | YES | |
| AML#14 | CEBPA | chr19:33792381 | -/CTT | c.939_940insAAG | p.K313dup | INDEL | Nonframe shift Insertion | 3 | 1 | YES (27.1) | YES (48.7) | N/E |
| CEBPA | chr19:33793174 | CG/C | c.146delC | p.P49fs | INDEL | Frameshift Deletion | 1 | 1 | YES (42.1) | YES (57.5) | N/E | |
| CEBPA | chr19:33792729 | G/A | c.592C > T | p.P198S | SNV | missense | 1 | 1 | NO | YES (16.2) | NO | |
| AML#15 | CEBPA | chr19:33792729 | G/A | c.592C > T | p.P198S | SNV | missense | 1 | 1 | NO | YES (14.8) | NO |
| AML#16 | NPM1 | chr5:170837545 | -/TGCA | c.861_862insTGCA | p.W288fs*12 | INDEL | Frameshift Insertion | 4 | 11 | YES (47.6) | YES (46.6) | N/E |
| IDH2 | chr15:90631934 | C/T | c.419G | p.R140 | SNV | missense | 1 | 4 | YES (60.7) | YES (53.8) | N/E | |
| AML#17 | NPM1 | chr5:170837545 | -/TGCA | c.861_862insTGCA | p.W288fs*12 | INDEL | Frameshift Insertion | 4 | 11 | YES (39.1) | YES (57.6) | N/E |
| FLT3 | chr13:28592642 | C/A | c.2503G | p.D835 | SNV | missense | 1 | 20 | YES (49.0) | YES (50.0) | N/E | |
| AML#18 | TP53 | chr17:7577120 | C/T | c.818G > A | p.R273H | SNV | missense | 1 | 8 | YES (78.5) | YES (72.8) | N/E |
| CEBPA | chr19:33792729 | G/A | c.592C > T | p.P198S | SNV | missense | 1 | 1 | NO | YES (15.7) | NO | |
| AML#19 | TP53 | chr17:7577538 | C/T | c.743G > A | p.R248Q | SNV | missense | 1 | 7 | YES (68.2) | YES (63.1) | N/E |
| AML#20 | TP53 | chr17:7577082 | C/T | c.856G > A | p.E286K | SNV | missense | 1 | 8 | YES (73.1) | YES (79.1) | N/E |
| AML#21 | TP53 | chr17:7577094 | G/A | c.844C > T | p.R282W | SNV | missense | 1 | 8 | YES (48.7) | YES (49.1) | N/E |
| TP53 | chr17:7578475 | G/C | c.455C > G | p.P152R | SNV | missense | 1 | 5 | YES (21.0) | YES (11.5) | N/E | |
| CEBPA | chr19:33792731 | -/GCGGGT | c.589_590insACCCGC | p.H195_P196dup | INDEL | Nonframe shift Insertion | 6 | 1 | NO | YES (25.2) | NO | |
| AML#22 | TP53 | chr17:7578413 | C/T | c.517G > A | p.V173M | SNV | missense | 1 | 5 | YES (18.1) | YES (45.3) | N/E |
| TP53 | chr17:7579715 | AG/A | c.81del | p.E28Kfs * 16 | INDEL | Frameshift Deletion | 1 | 3 | YES (45.0) | YES (41.8) | N/E |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cumbo, C.; Minervini, C.F.; Orsini, P.; Anelli, L.; Zagaria, A.; Minervini, A.; Coccaro, N.; Impera, L.; Tota, G.; Parciante, E.; et al. Nanopore Targeted Sequencing for Rapid Gene Mutations Detection in Acute Myeloid Leukemia. Genes 2019, 10, 1026. https://doi.org/10.3390/genes10121026
Cumbo C, Minervini CF, Orsini P, Anelli L, Zagaria A, Minervini A, Coccaro N, Impera L, Tota G, Parciante E, et al. Nanopore Targeted Sequencing for Rapid Gene Mutations Detection in Acute Myeloid Leukemia. Genes. 2019; 10(12):1026. https://doi.org/10.3390/genes10121026
Chicago/Turabian StyleCumbo, Cosimo, Crescenzio Francesco Minervini, Paola Orsini, Luisa Anelli, Antonella Zagaria, Angela Minervini, Nicoletta Coccaro, Luciana Impera, Giuseppina Tota, Elisa Parciante, and et al. 2019. "Nanopore Targeted Sequencing for Rapid Gene Mutations Detection in Acute Myeloid Leukemia" Genes 10, no. 12: 1026. https://doi.org/10.3390/genes10121026