Genomes of Three Closely Related Caribbean Amazons Provide Insight for Species History and Conservation

 , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
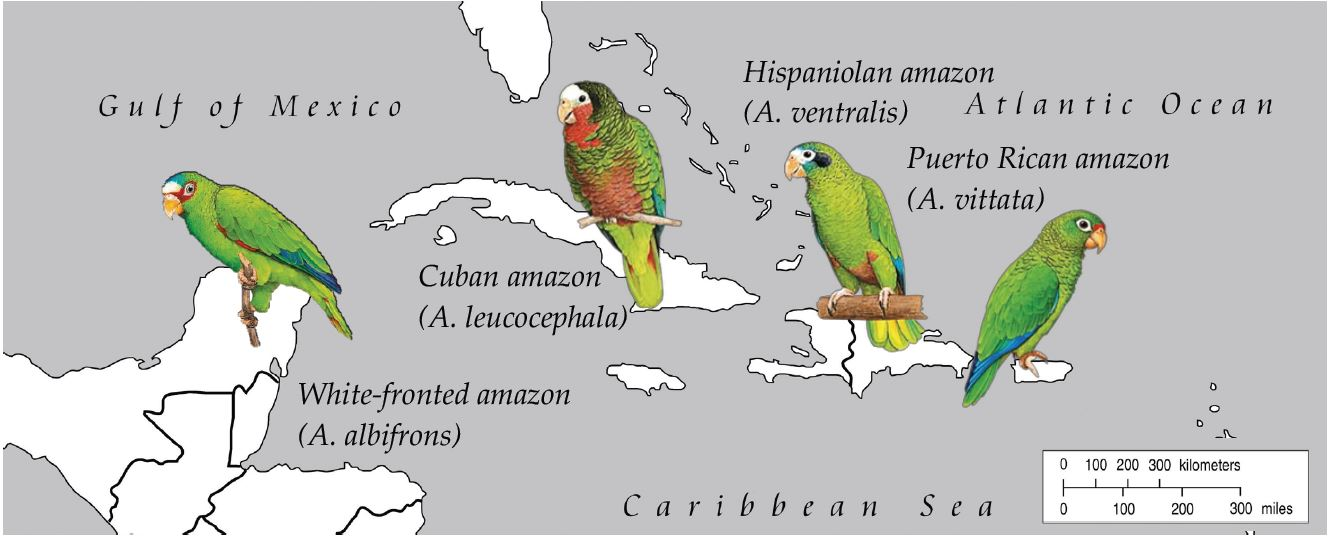
2.1. Samples
2.2. DNA and RNA Extraction
2.3. Genome and Transcriptome Sequencing
2.4. Data QC and Filtering
2.5. Genome and Transcriptome Assembly
2.6. Repeat Masking in the A. vittata Genome
2.7. Annotation of Protein-Coding Genes in the A. vittata Genome
2.8. Genome Read Alignment and Variant Calling
2.9. Reference-Assisted Assembly of A.ventralis and A. leucocephala Genomes
2.10. Phylogeny Reconstruction and Divergence Time Estimation
2.11. Demographic History Inference
2.12. Amazona Genome Browser Hub
3. Results
3.1. Assembly and Annotation
3.2. Genome-Wide Heterozygosity
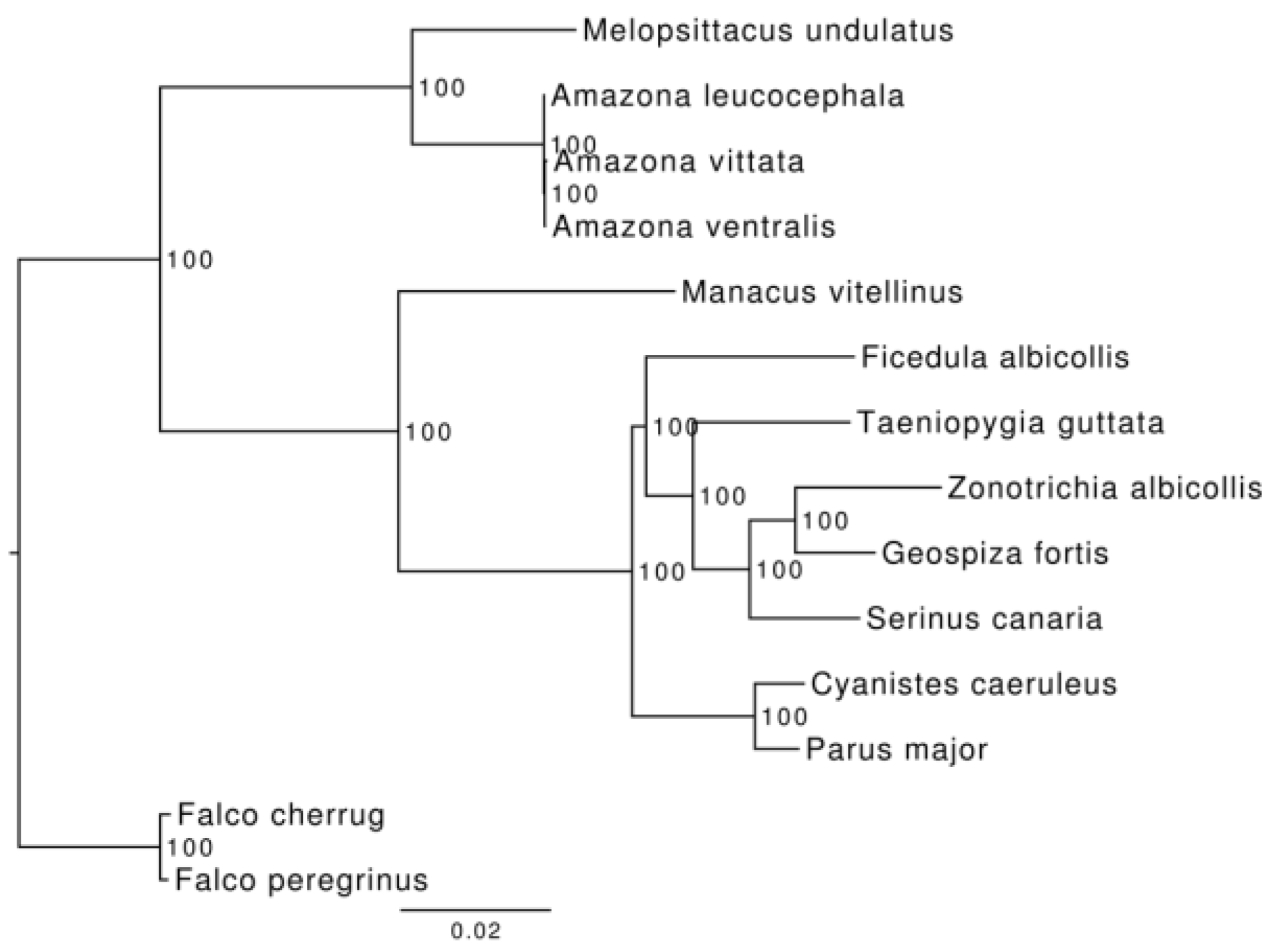
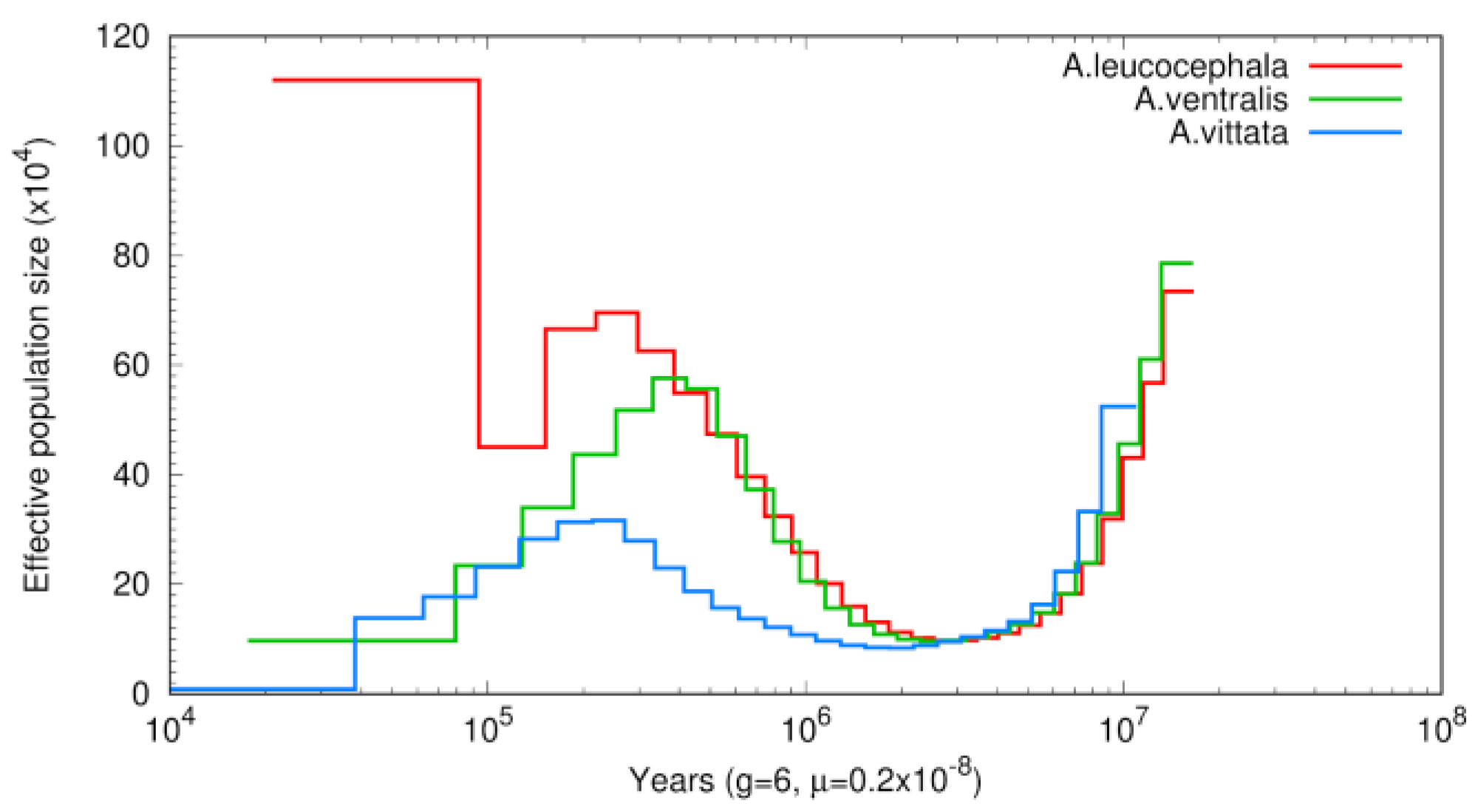
3.3. Phylogenetic and Demographic Analysis
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Zhang, G.; Jarvis, E.D.; Gilbert, M.T.P. A flock of Genomes. Science 2014, 346, 1308–1309. [Google Scholar] [CrossRef]
- Zhang, G. Genomics: Bird sequencing project takes off. Nature 2015, 522, 34. [Google Scholar] [CrossRef] [PubMed]
- Jarvis, E.D.; Mirarab, S.; Aberer, A.J.; Li, B.; Houde, P.; Li, C.; Ho, S.Y.W.; Faircloth, B.C.; Nabholz, B.; Howard, J.T.; et al. Whole-genome analyses resolve early branches in the tree of life of modern birds. Science 2014, 346, 1320–1331. [Google Scholar] [CrossRef] [Green Version]
- Zhang, G.; Li, C.; Li, Q.; Li, B.; Larkin, D.M.; Lee, C.; Storz, J.F.; Antunes, A.; Greenwold, M.J.; Meredith, R.W.; et al. Comparative genomics reveals insights into avian genome evolution and adaptation. Science 2014, 346, 1311–1320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grant, P.R.; Grant, B.R. Adaptive radiation of Darwin’s finches. Am. Sci. 2002. [Google Scholar] [CrossRef]
- MacArthur, R.H.; Wilson, E.O. The Theory of Island Biogeography, 1st ed.; Princeton University Press: Princeton, NJ, USA, 1967; ISBN 0691088365. [Google Scholar]
- Darwin, C. Journal of Researches into the Natural History and Geology of the Countries Visited during the Voyage of H.M.S. Beagle Round the World, under the Command of Capt. Fitz Roy, R.N. (Voyage on the Beagle), 2nd ed.; John Murray: London, UK, 1845. [Google Scholar]
- Whittaker, R.J.; Fernández-Palacios, J.M.; Matthews, T.J.; Borregaard, M.K.; Triantis, K.A. Island biogeography: Taking the long view of nature’s laboratories. Science 2017, 357, 6354. [Google Scholar] [CrossRef]
- O’Brien, S.J. Genome empowerment for the Puerto Rican parrot—Amazona vittata. Gigascience 2012, 1, 13. [Google Scholar] [CrossRef] [PubMed]
- Snyder, N.; Wiley, J.W.; Kepler, C.B. The Parrots of Luquillo: Natural History and Conservation of the Puerto Rican Parrot; Western Foundation of Vertebrate Zoology: Los Angeles, CA, USA, 1987. [Google Scholar]
- Lack, D. Island Biology Illustrated by the Land Birds of Jamaica; Blackwell: Oxford, UK, 1976. [Google Scholar]
- Russello, M.A.; Amato, G. A molecular phylogeny of Amazona: Implications for Neotropical parrot biogeography, taxonomy, and conservation. Mol. Phylogenet. Evol. 2004, 30, 421–437. [Google Scholar] [CrossRef]
- Blanco, G.; Hiraldo, F.; Rojas, A.; Dénes, F.V.; Tella, J.L. Parrots as key multilinkers in ecosystem structure and functioning. Ecol. Evol. 2015, 5, 4141–4160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blanco, G.; Hiraldo, F.; Tella, J.L. Ecological functions of parrots: An integrative perspective from plant life cycle to ecosystem functioning. Emu Aust. Ornithol. 2018, 118, 36–49. [Google Scholar] [CrossRef]
- Aslan, C.E.; Zavaleta, E.S.; Croll, D.; Tershy, B. Effects of Native and Non-Native Vertebrate Mutualists on Plants. Conserv. Biol. 2012, 26, 778–789. [Google Scholar] [CrossRef]
- Anderson, S.H.; Kelly, D.; Ladley, J.J.; Molloy, S.; Terry, J. Cascading effects of bird functional extinction reduce pollination and plant density. Science 2011, 331, 1068–1071. [Google Scholar] [CrossRef]
- Cracraft, J. Avian evolution, Gondwana biogeography and the Cretaceous-Tertiary mass extinction event. Proc. R. Soc. B Biol. Sci. 2001, 268, 459–469. [Google Scholar] [CrossRef]
- Wright, T.F.; Schirtzinger, E.E.; Matsumoto, T.; Eberhard, J.R.; Graves, G.R.; Sanchez, J.J.; Capelli, S.; Müller, H.; Scharpegge, J.; Chambers, G.K.; et al. A multilocus molecular phylogeny of the parrots (Psittaciformes): Support for a gondwanan origin during the cretaceous. Mol. Biol. Evol. 2008, 25, 2141–2156. [Google Scholar] [CrossRef] [PubMed]
- Rheindt, F.E.; Christidis, L.; Kuhn, S.; de Kloet, S.; Norman, J.A.; Fidler, A. The timing of diversification within the most divergent parrot clade. J. Avian Biol. 2014, 45, 140–148. [Google Scholar] [CrossRef]
- Prum, R.O.; Berv, J.S.; Dornburg, A.; Field, D.J.; Townsend, J.P.; Moriarty Lemmon, E.; Lemmon, A.R. A Comprehensive Phylogeny of Birds (Aves) using Targeted Next Generation DNA Sequencing Online Data and Software Archive. Nature 2015, 526, 569–573. [Google Scholar] [CrossRef] [PubMed]
- Claramunt, S.; Cracraft, J. A new time tree reveals Earth history’s imprint on the evolution of modern birds. Sci. Adv. 2015, 564, 136–141. [Google Scholar] [CrossRef]
- Mayr, G. The origins of crown group birds: Molecules and fossils. Palaeontology 2014, 57, 231–242. [Google Scholar] [CrossRef]
- Mayr, G. Paleogene Fossil Birds; Springer-Verlag: Berlin/Heidelberg, Germany, 2009. [Google Scholar]
- Ottens-Wainright, P.; Halanych, K.M.; Eberhard, J.R.; Burke, R.I.; Wiley, J.W.; Gnam, R.S.; Aquilera, X.G. Independent geographic origin of the genus Amazona in the West Indies. J. Caribb. Ornithol. 2004, 17, 23–49. [Google Scholar]
- Avise, J.C. Phylogeography: The History and Formation of Species; Harvard University Press: Boston, MA, USA, 2000. [Google Scholar]
- Kolchanova, S. Molecular Phylogeny and Evolution of Amazon Parrots in the Greater Antilles; Univeristy of Puerto Rico at Mayaguez: Mayagüez, Puerto Rico, 2018. [Google Scholar]
- Oleksyk, T.K.; Pombert, J.-F.; Siu, D.; Mazo-Vargas, A.; Ramos, B.; Guiblet, W.; Afanador, Y.; Ruiz-Rodriguez, C.T.; Nickerson, M.L.; Logue, D.M.; et al. A locally funded Puerto Rican parrot (Amazona vittata) genome sequencing project increases avian data and advances young researcher education. Gigascience 2012, 1, 14. [Google Scholar] [CrossRef] [Green Version]
- Brinkley, D. The Wilderness Warrior: Theodore Roosevelt and the Crusade for America; Harper Collins: New York, NY, USA, 2009. [Google Scholar]
- Afanador, Y.; Velez-Valentín, J.; Valentín de la Rosa, R.; Martínez-Cruzado, J.C.; vonHoldt, B.; Oleksyk, K.T. Isolation and characterization of microsatellite loci in the critically endangered Puerto Rican parrot (Amazona vittata). Conserv. Genet. Resour. 2014, 6, 885–889. [Google Scholar] [CrossRef]
- Brock, M.K.; White, B.N. Application of DNA fingerprinting to the recovery program of the endangered Puerto Rican parrot. Proc. Natl. Acad. Sci. USA 1992, 89, 11121–11125. [Google Scholar] [CrossRef] [PubMed]
- Allendorf, F.W.; Hohenlohe, P.A.; Luikart, G. Genomics and the future of conservation genetics. Nat. Rev. Genet. 2010, 11, 697–709. [Google Scholar] [CrossRef] [PubMed]
- Ouborg, N.J.; Pertoldi, C.; Loeschcke, V.; Bijlsma, R.K.; Hedrick, P.W. Conservation genetics in transition to conservation genomics. Trends Genet. 2010, 26, 177–187. [Google Scholar] [CrossRef] [PubMed]
- Andrews, S. FASTQC. A Quality Control Tool for High Throughput Sequence Data. 2010. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 15 July 2019).
- Starostina, E.; Tamazian, G.; Dobrynin, P.; O’Brien, S.; Komissarov, A. Cookiecutter: A tool for kmer-based read filtering and extraction. bioRxiv 2015, 2015, 24679. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Trinity: Recontructing a full-length transcriptome assembly without a genome from RNA-Seq data. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef]
- Grigorev, K.; Kliver, S.; Dobrynin, P.; Komissarov, A.; Wolfsberger, W.; Krasheninnikova, K.; Afanador-Hernández, Y.M.; Paulino, L.A.; Carreras, R.; Rodríguez, L.E.; et al. Innovative assembly strategy contributes to the understanding of evolution and conservation genetics of the critically endangered Solenodon paradoxus from the island of Hispaniola. GigaScience 2018, 7, giy025. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- Boetzer, M.; Henkel, C.V.; Jansen, H.J.; Butler, D.; Pirovano, W. Scaffolding pre-assembled contigs using SSPACE. Bioinformatics 2011, 27, 578–579. [Google Scholar] [CrossRef]
- Luo, R.; Liu, B.; Xie, Y.; Li, Z.; Huang, W.; Yuan, J.; He, G.; Chen, Y.; Pan, Q.; Liu, Y.; et al. SOAPdenovo2: An empirically improved memory-efficient short-read de novo assembler. Gigascience 2012, 1, 18. [Google Scholar] [CrossRef]
- Kent, W.J. BLAT—The BLAST-like alignment tool. Genome Res. 2002, 12, 656–664. [Google Scholar] [CrossRef] [PubMed]
- Xue, W.; Li, J.T.; Zhu, Y.P.; Hou, G.Y.; Kong, X.F.; Kuang, Y.Y.; Sun, X.W. L_RNA_scaffolder: Scaffolding genomes with transcripts. BMC Genom. 2013, 14, 604. [Google Scholar] [CrossRef] [PubMed]
- Marçais, G.; Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 2011, 27, 764–770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kliver, S. KRATeR. Available online: https://github.com/mahajrod/KrATER (accessed on 15 July 2019).
- Simão, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef] [PubMed]
- Bao, W.; Kojima, K.K.; Kohany, O. Repbase Update, a database of repetitive elements in eukaryotic genomes. Mob. DNA 2015, 6, 11. [Google Scholar] [CrossRef]
- Smit, A.; Hubley, R.; Green, P. RepeatMasker Open-4.0. 2013–2018. Available online: http://www.repeatmasker.org (accessed on 10 June 2018).
- Smit, A.; Hubley, R. RepeatModeler Open-1.0. Available online: https://github.com/rmhubley/RepeatModeler/blob/master/README (accessed on 14 July 2018).
- Tarailo-Graovac, M.; Chen, N. Using RepeatMasker to identify repetitive elements in genomic sequences. Curr. Protoc. Bioinform. 2009, 5, 4–10. [Google Scholar] [CrossRef]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef]
- Slater, G.S.C.; Birney, E. Automated generation of heuristics for biological sequence comparison. BMC Bioinformatics 2005, 6, 31. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Stanke, M.; Keller, O.; Gunduz, I.; Hayes, A.; Waack, S.; Morgenstern, B. AUGUSTUS: Ab initio prediction of alternative transcripts. Nucleic Acids Res. 2006, 34, W435–W439. [Google Scholar] [CrossRef] [PubMed]
- Johnson, L.S.; Eddy, S.R.; Portugaly, E. Hidden Markov model speed heuristic and iterative HMM search procedure. BMC Bioinform. 2010, 11, 431. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Bateman, A.; Coin, L.; Durbin, R.; Finn, R.D.; Hollich, V.; Griffiths-Jones, S.; Khanna, A.; Marshall, M.; Moxon, S.; Sonnhammer, E.L.L.; et al. The Pfam protein families database. Nucleic Acids Res. 2004, 32, D138–D141. [Google Scholar] [CrossRef] [PubMed]
- Bateman, A.; Martin, M.J.; O’Donovan, C.; Magrane, M.; Alpi, E.; Antunes, R.; Bely, B.; Bingley, M.; Bonilla, C.; Britto, R.; et al. UniProt: The universal protein knowledgebase. Nucleic Acids Res. 2017, 45, D158–D169. [Google Scholar] [CrossRef]
- Van der Auwera, G.A.; Carneiro, M.O.; Hartl, C.; Poplin, R.; del Angel, G.; Levy-Moonshine, A.; Jordan, T.; Shakir, K.; Roazen, D.; Thibault, J.; et al. From fastQ data to high-confidence variant calls: The genome analysis toolkit best practices pipeline. Curr. Protoc. Bioinform. 2013, 43, 11.10.1–11.10.33. [Google Scholar] [CrossRef]
- Huerta-Cepas, J.; Szklarczyk, D.; Forslund, K.; Cook, H.; Heller, D.; Walter, M.C.; Rattei, T.; Mende, D.R.; Sunagawa, S.; Kuhn, M.; et al. eggNOG 4.5: A hierarchical orthology framework with improved functional annotations for eukaryotic, prokaryotic and viral sequences. Nucleic Acids Res. 2015, 44, gkv1248. [Google Scholar] [CrossRef] [PubMed]
- Frankl-Vilches, C.; Kuhl, H.; Werber, M.; Klages, S.; Kerick, M.; Bakker, A.; de Oliveira, E.H.C.; Reusch, C.; Capuano, F.; Vowinckel, J.; et al. Using the canary genome to decipher the evolution of hormone-sensitive gene regulation in seasonal singing birds. Genome Biol. 2015, 16, 19. [Google Scholar] [CrossRef]
- Ellegren, H.; Smeds, L.; Burri, R.; Olason, P.I.; Backström, N.; Kawakami, T.; Künstner, A.; Mäkinen, H.; Nadachowska-Brzyska, K.; Qvarnström, A.; et al. The genomic landscape of species divergence in Ficedula flycatchers. Nature 2012, 491, 756. [Google Scholar] [CrossRef]
- Laine, V.N.; Gossmann, T.I.; Schachtschneider, K.M.; Garroway, C.J.; Madsen, O.; Verhoeven, K.J.F.; de Jager, V.; Megens, H.-J.; Warren, W.C.; Minx, P.; et al. Evolutionary signals of selection on cognition from the great tit genome and methylome. Nat. Commun. 2016, 7, 10474. [Google Scholar] [CrossRef] [Green Version]
- Balakrishnan, C.N.; Mukai, M.; Gonser, R.A.; Wingfield, J.C.; London, S.E.; Tuttle, E.M.; Clayton, D.F. Brain transcriptome sequencing and assembly of three songbird model systems for the study of social behavior. PeerJ 2014, 2, e396. [Google Scholar] [CrossRef] [PubMed]
- Mueller, J.C.; Kuhl, H.; Timmermann, B.; Kempenaers, B. Characterization of the genome and transcriptome of the blue tit Cyanistes caeruleus: Polymorphisms, sex-biased expression and selection signals. Mol. Ecol. Resour. 2016, 16, 549–561. [Google Scholar] [CrossRef]
- Ganapathy, G.; Howard, J.T.; Ward, J.M.; Li, J.; Li, B.; Li, Y.; Xiong, Y.; Zhang, Y.; Zhou, S.; Schwartz, D.C.; et al. High-coverage sequencing and annotated assemblies of the budgerigar genome. Gigascience 2014, 3, 2011–2047. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Parker, P.; Li, B.; Li, H.; Wang, J.; Parker, P.; Li, B.; Li, H.; Wang, J. The genome of Darwin’s Finch (Geospiza fortis). GigaDB. 2012. Available online: http://dx.doi.org/10.5524/100040 (accessed on 10 June 2018).
- Warren, W.C.; Clayton, D.F.; Ellegren, H.; Arnold, A.P.; Hillier, L.W.; Künstner, A.; Searle, S.; White, S.; Vilella, A.J.; Fairley, S.; et al. The genome of a songbird. Nature 2010, 464, 757–762. [Google Scholar] [CrossRef] [PubMed]
- Doyle, J.M.; Katzner, T.E.; Bloom, P.H.; Ji, Y.; Wijayawardena, B.K.; DeWoody, J.A. The Genome Sequence of a Widespread Apex Predator, the Golden Eagle (Aquila chrysaetos). PLoS ONE 2014, 9, e95599. [Google Scholar] [CrossRef] [PubMed]
- Zhan, X.; Pan, S.; Wang, J.; Dixon, A.; He, J.; Muller, M.G.; Ni, P.; Hu, L.; Liu, Y.; Hou, H.; et al. Peregrine and saker falcon genome sequences provide insights into evolution of a predatory lifestyle. Nat. Genet. 2013, 45, 563. [Google Scholar] [CrossRef]
- Löytynoja, A.; Goldman, N. webPRANK: A phylogeny-aware multiple sequence aligner with interactive alignment browser. BMC Bioinform. 2010, 11, 579. [Google Scholar] [CrossRef]
- Talavera, G.; Castresana, J. Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst. Biol. 2007, 56, 564–577. [Google Scholar] [CrossRef] [PubMed]
- Castresana, J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol. Biol. Evol. 2000, 17, 540–552. [Google Scholar] [CrossRef]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef]
- Rambaut, A. FigTree. 2016. Available online: http://tree.bio.ed.ac.uk/software/figtree/ (accessed on 10 June 2018).
- Li, H.; Durbin, R. Inference of human population history from individual whole-genome sequences. Nature 2011, 475, 493–496. [Google Scholar] [CrossRef] [Green Version]
- Earnhardt, J.; Vélez-Valentín, J.; Valentin, R.; Long, S.; Lynch, C.; Schowe, K. The Puerto Rican parrot reintroduction program: Sustainable management of the aviary population. Zoo Biol. 2014, 33, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Smeds, L.; Qvarnström, A.; Ellegren, H. Direct estimate of the rate of germline mutation in a bird. Genome Res. 2016, 6, 1211–1218. [Google Scholar] [CrossRef] [PubMed]
- Raney, B.J.; Dreszer, T.R.; Barber, G.P.; Clawson, H.; Fujita, P.A.; Wang, T.; Nguyen, N.; Paten, B.; Zweig, A.S.; Karolchik, D.; et al. Track data hubs enable visualization of user-defined genome-wide annotations on the UCSC Genome Browser. Bioinformatics 2014, 30, 1003–1005. [Google Scholar] [CrossRef] [PubMed]
- Kent, W.J.; Zweig, A.S.; Barber, G.; Hinrichs, A.S.; Karolchik, D. BigWig and BigBed: Enabling browsing of large distributed datasets. Bioinformatics 2010, 26, 2204–2207. [Google Scholar] [CrossRef] [PubMed]
- Tiersch, T.R.; Wachtel, S.S. On the evolution of genome size of birds. J. Hered. 1991, 82, 363–368. [Google Scholar] [CrossRef]
- Gregory, T. Animal Genome Size Database. Available online: http://www.genomesize.com (accessed on 15 July 2018).
- Volfovsky, N.; Oleksyk, T.K.; Cruz, K.C.; Truelove, A.L.; Stephens, R.M.; Smith, M.W. Chimpanzee chromosome 23 vs. human 22: Genomic insertion, deletion and ancestral indel polymorphisms. BMC Genom. 2009, 10. [Google Scholar] [CrossRef]
- Organ, C.L.; Shedlock, A.M.; Meade, A.; Pagel, M.; Edwards, S.V. Origin of avian genome size and structure in non-avian dinosaurs. Nature 2007, 446, 180. [Google Scholar] [CrossRef]
- Kapusta, A.; Suh, A.; Feschotte, C. Dynamics of genome size evolution in birds and mammals. Proc. Natl. Acad. Sci. USA 2017. [Google Scholar] [CrossRef]
- Coullin, P.; Bed’Hom, B.; Candelier, J.J.; Vettese, D.; Maucolin, S.; Moulin, S.; Galkina, S.A.; Bernheim, A.; Volobouev, V. Cytogenetic repartition of chicken CR1 sequences evidenced by PRINS in Galliformes and some other birds. Chromosom. Res. 2005, 13, 665–673. [Google Scholar] [CrossRef]
- Ksepka, D.; Clarke, J. Phylogenetically vetted and stratigraphically constrained fossil calibrations within Aves. Palaeontol. Electron. 2015, 18, 1–25. [Google Scholar] [CrossRef]
- DeWoody, Y.D.; DeWoody, J.A. On the estimation of genome-wide heterozygosity using molecular markers. J. Hered. 2005, 96, 85–88. [Google Scholar] [CrossRef] [PubMed]
- Coltman, D.W.; Slate, J. Microsatellite measures of inbreeding: A meta-analysis. Evolution 2003, 57, 971–983. [Google Scholar] [CrossRef]
- Chapman, J.R.; Nakagawa, S.; Coltman, D.W.; Slate, J.; Sheldon, B.C. A quantitative review of heterozygosity-fitness correlations in animal populations. Mol. Ecol. 2009. [Google Scholar] [CrossRef]
- Spielman, D.; Brook, B.W.; Frankham, R. Most species are not driven to extinction before genetic factors impact them. Proc. Natl. Acad. Sci. USA 2004, 101, 15261–15264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vellend, M. Island Biogeography of Genes and Species. Am. Nat. 2003, 162, 358–365. [Google Scholar] [CrossRef] [PubMed]
- Wright, S. Breeding Structure of Populations in Relation to Speciation. Am. Nat. 1940, 74, 232–248. [Google Scholar] [CrossRef]
- Fleming, T.H. The theory of island biogeography at age 40. Evolution 2010. [Google Scholar] [CrossRef]
- Li, S.; Li, B.; Cheng, C.; Xiong, Z.; Liu, Q.; Lai, J.; Carey, H.V.; Zhang, Q.; Zheng, H.; Wei, S.; et al. Genomic signatures of near-extinction and rebirth of the crested ibis and other endangered bird species. Genome Biol. 2014, 15, 557. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Parrot Species | PE Library Coverage | Genome Size (Gbp) | C-Value (pg) |
|---|---|---|---|
| Amazona vitatta | 14× | 1.42 | 1.58 |
| A. ventralis | 22× | 1.42 | 1.62–1.65 |
| A. leucocephala | 16× | 1.54 | 1.58–1.65 |
| Library ID | Tissue | Read Pairs (Millions) | Bases (Gbp) | Assembled Transcripts |
|---|---|---|---|---|
| Parrot13 | Blood | 54.2 | 10.7 | 314,505 |
| Parrot140 | Blood | 64.9 | 12.7 | 378,318 |
| Parrot335 | Blood | 47.3 | 9.3 | 306,123 |
| Parrot341 | Blood | 54.2 | 10.7 | 326,706 |
| Parrot_336 | Liver | 69.6 | 13.7 | 210,549 |
| Merged | - | 290.3 | 57.1 | 680,785 |
| N50 (kbp) | L50 (kbp) | Longest Contig (Mb) | Number of Ns Mb | Number of Scaffolds | Assembled Genome Length (Gbp) | |
|---|---|---|---|---|---|---|
| Amazona vittata | 101.028 | 3057 | 1.885 | 367.97 | 62,777 | 1.447 |
| Class | Number of Repeats | Total Length (bp) | Percentage of the Genome (%) |
|---|---|---|---|
| Total repeats: | 107,498,949 | 7.43% | |
| SINEs | 6995 | 895,617 | 0.06% |
| ALUs | 0 | 0 | 0.00% |
| MIRs | 3794 | 414,542 | 0.03% |
| LINEs: | 147,387 | 36,382,264 | 2.51% |
| LINE1 | 80 | 19,168 | 0.00% |
| LINE2 | 2175 | 474,752 | 0.03% |
| L3/CR1 | 144,820 | 35,707,674 | 2.47% |
| LTR elements | 34,688 | 10,514,590 | 0.73% |
| ERVL | 31,743 | 9,376,828 | 0.65% |
| ERVL-MaLRs | 0 | 0 | 0.00% |
| ERV_classI | 1826 | 779,161 | 0.05% |
| ERV_classII | 939 | 326,675 | 0.02% |
| DNA elements: | 19,273 | 3,034,179 | 0.21% |
| hAT-Charlie | 201 | 57,997 | 0.00% |
| TcMar-Tigger | 273 | 49,514 | 0.00% |
| Unclassified: | 345,805 | 56,672,299 | 3.92% |
| Small RNA: | 2066 | 272,961 | 0.02% |
| Satellites: | 3155 | 513,361 | 0.04% |
| Simple repeats: | 8207 | 1,700,409 | 0.12% |
| Low complexity: | 256 | 63,286 | 0.00% |
| Species | N of Genes | N of Genes with Longest Protein <100 aa | N Genes Assigned to the EggNOG Clusters | Genome Size (pg) * |
|---|---|---|---|---|
| Cyanistes caeruleus | 16,519 | 503 | 16,030 | 1.47 |
| Falco cherrug | 14,694 | 302 | 14,607 | - |
| Falco peregrinus | 14,859 | 307 | 14,771 | 1.45 |
| Ficedula albicollis | 15,400 | 360 | 14,952 | - |
| Geospiza fortis | 14,182 | 327 | 14,101 | - |
| Manacus vitellinus | 16,312 | 362 | 16,086 | - |
| Melopsittacus undullatus | 14,255 | 315 | 14,192 | 1.02–1.37 |
| Parus major | 15,251 | 285 | 14,795 | 1.51 |
| Serinus canaria | 15,582 | 455 | 15,194 | 1.48–1.62 |
| Taenopygia guttata | 16,368 | 494 | 16,202 | 1.25 |
| Zonotrichia albicollis | 14,374 | 314 | 14,018 | 1.33–1.58 |
| Amazona vittata | 19,669 | 2339 | 18,488 | 1.58 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kolchanova, S.; Kliver, S.; Komissarov, A.; Dobrinin, P.; Tamazian, G.; Grigorev, K.; Wolfsberger, W.W.; Majeske, A.J.; Velez-Valentin, J.; Valentin de la Rosa, R.; et al. Genomes of Three Closely Related Caribbean Amazons Provide Insight for Species History and Conservation. Genes 2019, 10, 54. https://doi.org/10.3390/genes10010054
Kolchanova S, Kliver S, Komissarov A, Dobrinin P, Tamazian G, Grigorev K, Wolfsberger WW, Majeske AJ, Velez-Valentin J, Valentin de la Rosa R, et al. Genomes of Three Closely Related Caribbean Amazons Provide Insight for Species History and Conservation. Genes. 2019; 10(1):54. https://doi.org/10.3390/genes10010054
Chicago/Turabian StyleKolchanova, Sofiia, Sergei Kliver, Aleksei Komissarov, Pavel Dobrinin, Gaik Tamazian, Kirill Grigorev, Walter W. Wolfsberger, Audrey J. Majeske, Jafet Velez-Valentin, Ricardo Valentin de la Rosa, and et al. 2019. "Genomes of Three Closely Related Caribbean Amazons Provide Insight for Species History and Conservation" Genes 10, no. 1: 54. https://doi.org/10.3390/genes10010054