The NLRP3 Inflammasome Role in the Pathogenesis of Pregnancy Induced Hypertension and Preeclampsia



Abstract
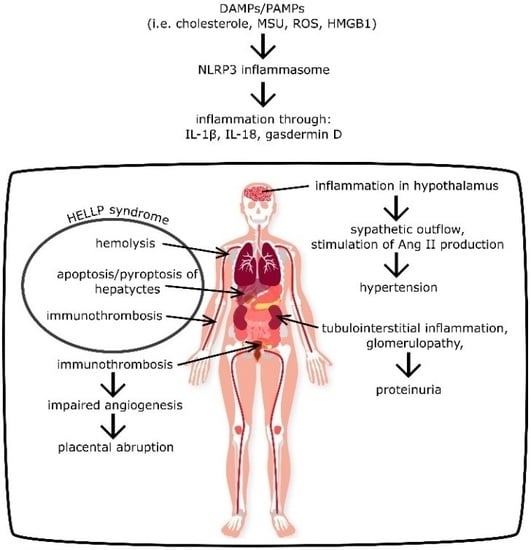
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Role of NLRP3 in PE and PIH
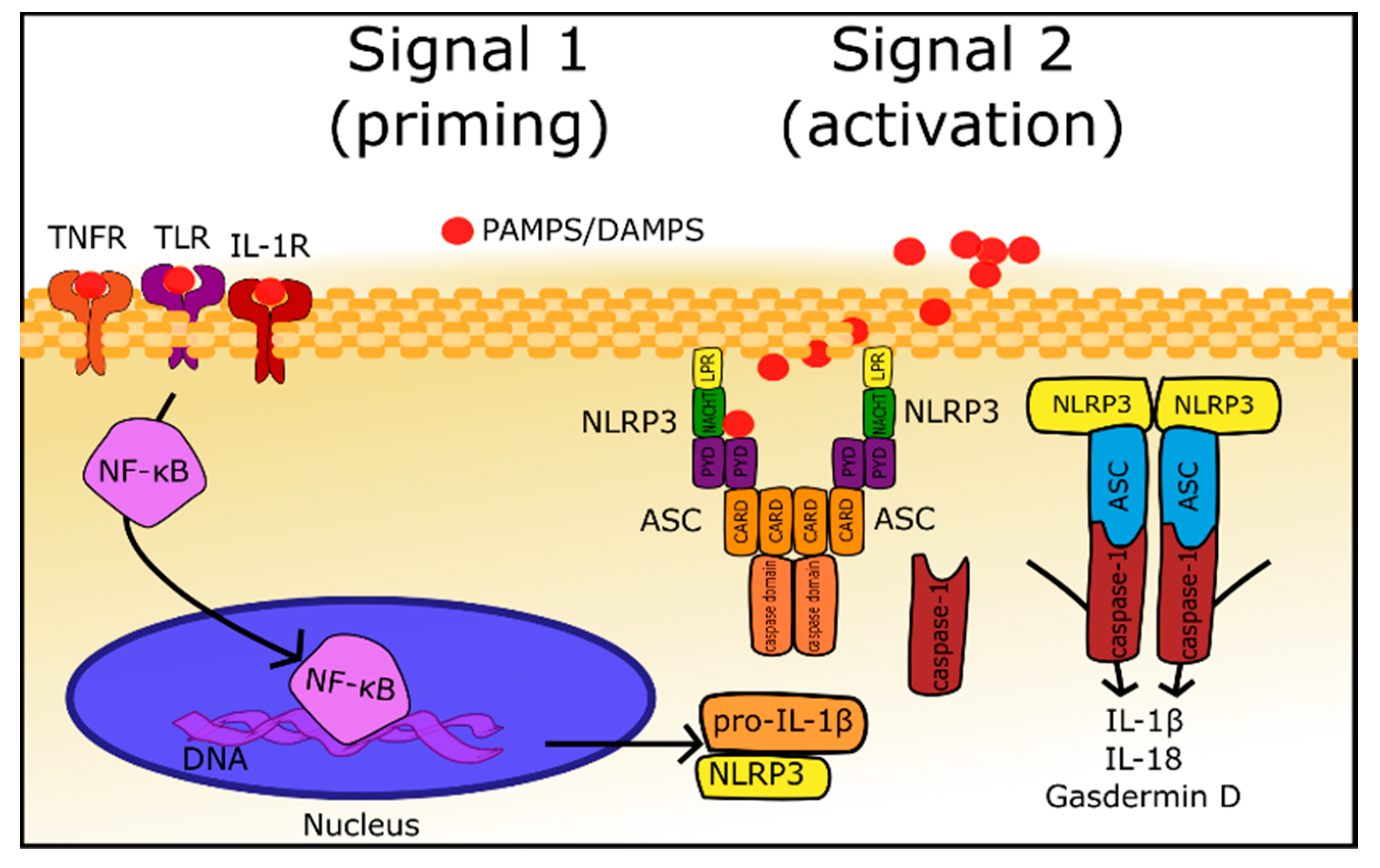
2.1. General Information
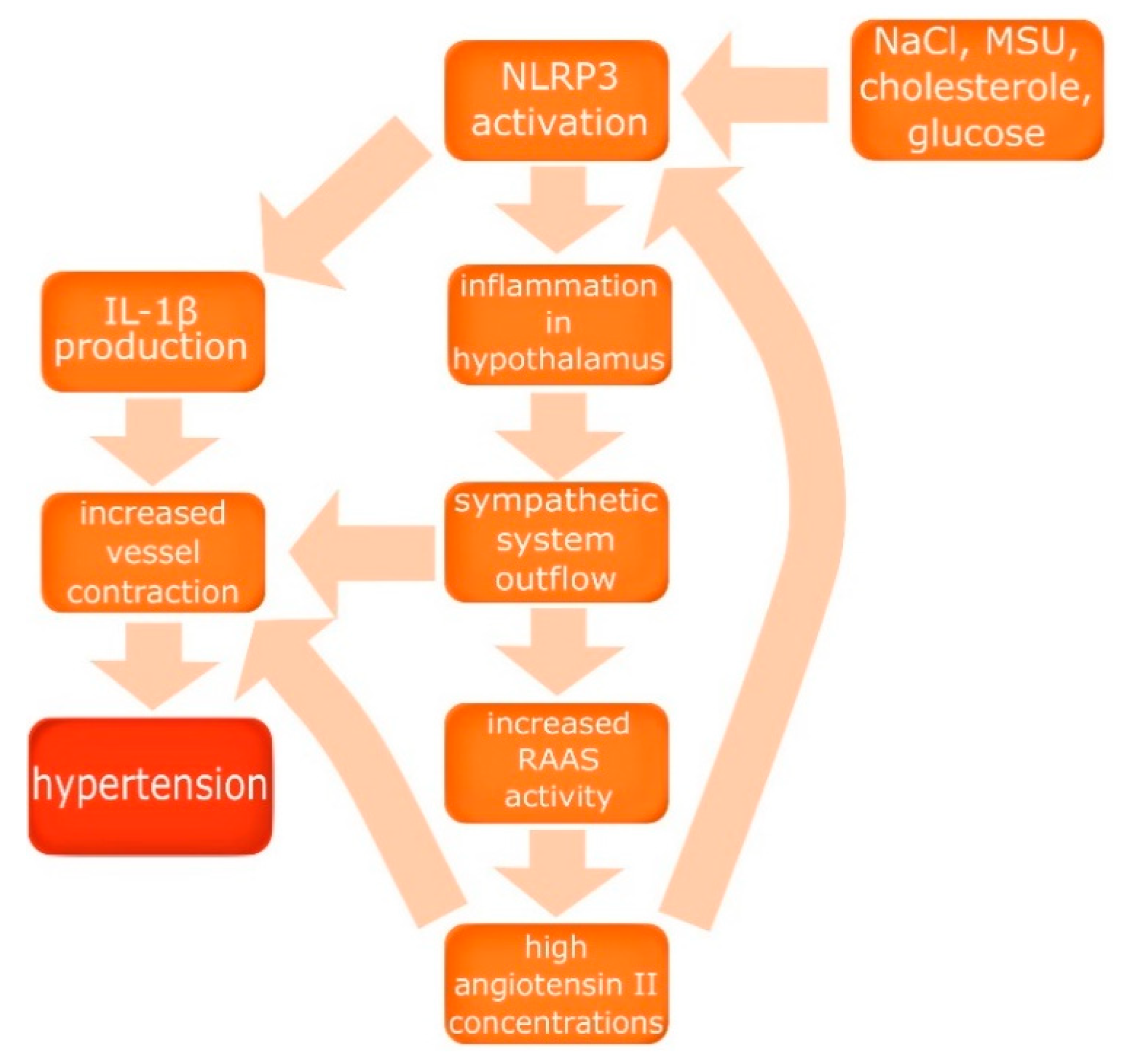
2.2. NLRP3 as an Inductor of Hypertension
2.3. Role of NLRP3 in Kidney Injury
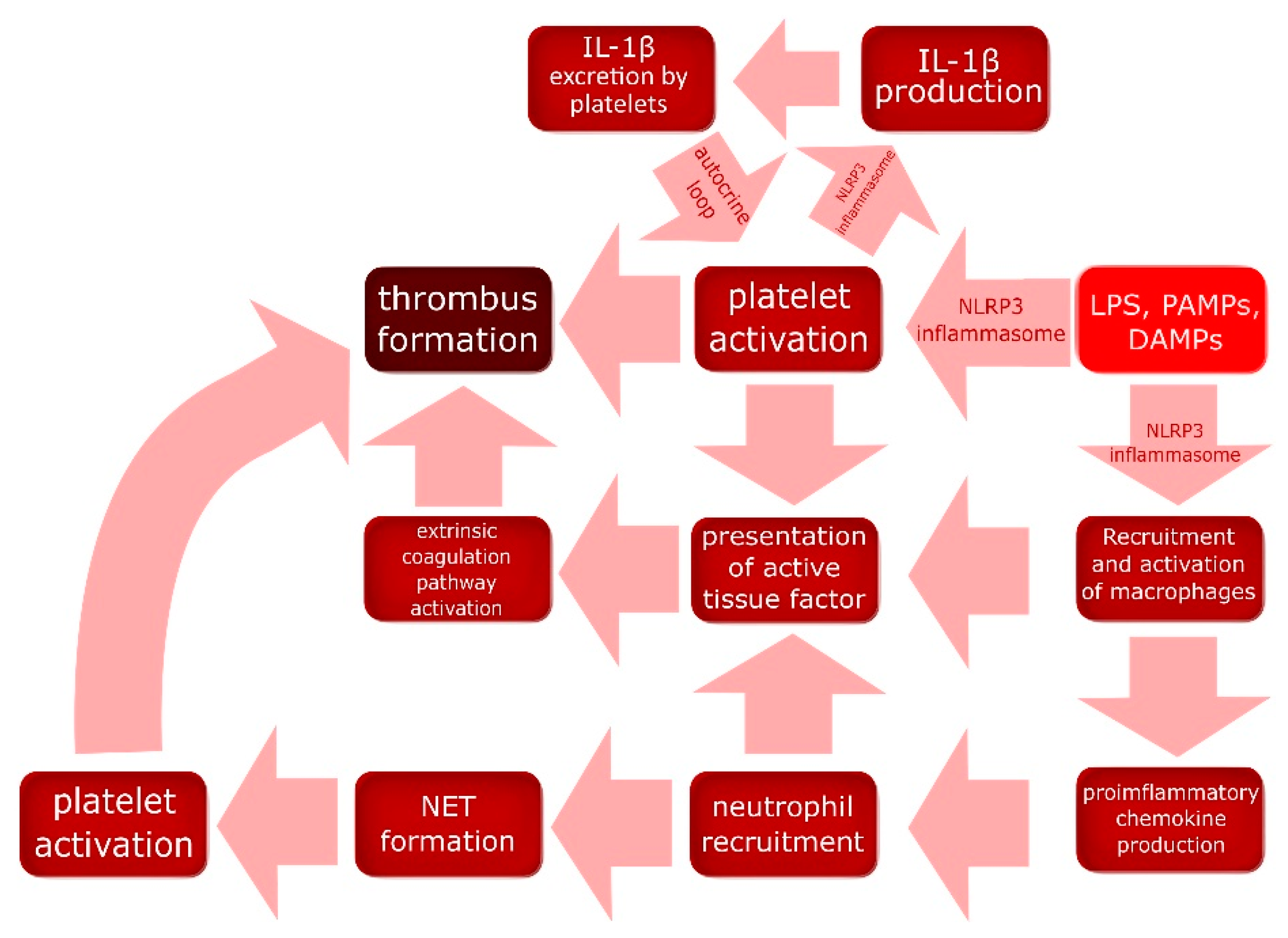
2.4. NLRP3 Impact on the Coagulation System
2.5. NLRP3 as an Inductor of Placental Abruption
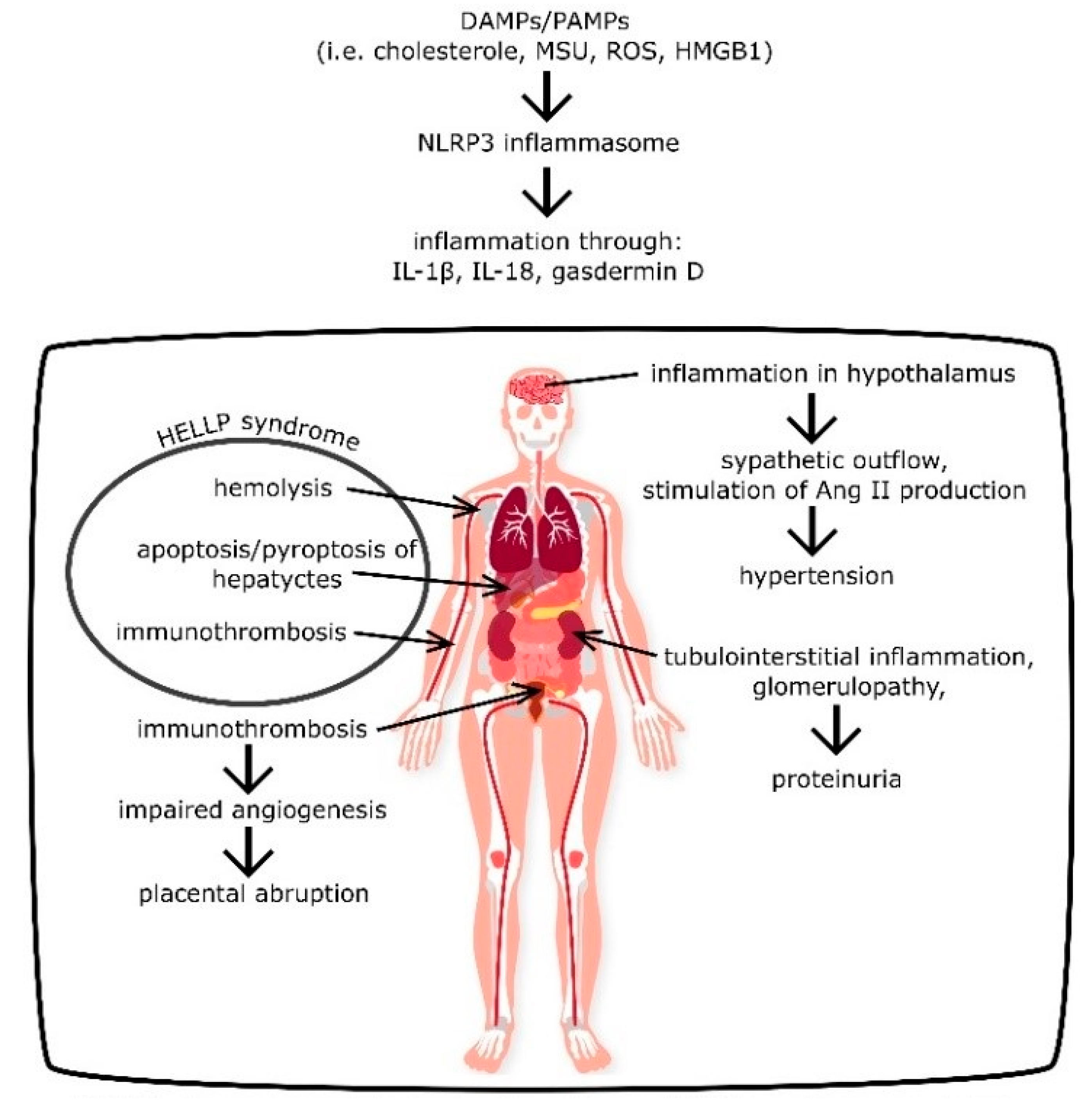
2.6. NLRP3 as an Inductor of HELLP Syndrome
3. NLRP3 Activators in PE and PIH
4. Outstanding Issues and Directions of Future Development
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kintiraki, E.; Papakatsika, S.; Kotronis, G.; Goulis, D.G.; Kotsis, V. Pregnancy-Induced hypertension. Horm. Athens Greece 2015, 14, 211–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghulmiyyah, L.; Sibai, B. Maternal Mortality from Preeclampsia/Eclampsia. Semin. Perinatol. 2012, 36, 56–59. [Google Scholar] [CrossRef]
- Weel, I.C.; Romão-Veiga, M.; Matias, M.L.; Fioratti, E.G.; Peraçoli, J.C.; Borges, V.T.; Araujo, J.P.; Peraçoli, M.T. Increased expression of NLRP3 inflammasome in placentas from pregnant women with severe preeclampsia. J. Reprod. Immunol. 2017, 123, 40–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, L.; Li, S.; Liu, Z.; Jiang, S.; Wang, J.; Guo, M.; Zhao, X.; Song, W.; Liu, S. The NLRP3 rs10754558 polymorphism is a risk factor for preeclampsia in a Chinese Han population. J. Matern. Fetal Neonatal Med. 2019, 32, 1792–1799. [Google Scholar] [CrossRef] [PubMed]
- Pontillo, A.; Reis, E.C.; Bricher, P.N.; Vianna, P.; Diniz, S.; Fernandes, K.S.; Chies, J.A.; Sandrim, V. NLRP1 L155H Polymorphism is a Risk Factor for Preeclampsia Development. Am. J. Reprod. Immunol. 2015, 73, 577–581. [Google Scholar] [CrossRef]
- Kelley, N.; Jeltema, D.; Duan, Y.; He, Y. The NLRP3 Inflammasome: An Overview of Mechanisms of Activation and Regulation. Int. J. Mol. Sci. 2019, 20, 3328. [Google Scholar] [CrossRef] [Green Version]
- Sutterwala, F.S.; Haasken, S.; Cassel, S.L. Mechanism of NLRP3 inflammasome activation. Ann. N. Y. Acad. Sci. 2014, 1319, 82–95. [Google Scholar] [CrossRef]
- Liu, D.; Zeng, X.; Li, X.; Mehta, J.L.; Wang, X. Role of NLRP3 inflammasome in the pathogenesis of cardiovascular diseases. Basic Res. Cardiol. 2017, 113, 5. [Google Scholar] [CrossRef]
- Krishnan, S.M.; Sobey, C.G.; Latz, E.; Mansell, A.; Drummond, G.R. IL-1β and IL-18: Inflammatory markers or mediators of hypertension? Br. J. Pharmacol. 2014, 171, 5589–5602. [Google Scholar] [CrossRef]
- Omi, T.; Kumada, M.; Kamesaki, T.; Okuda, H.; Munkhtulga, L.; Yanagisawa, Y.; Utsumi, N.; Gotoh, T.; Hata, A.; Soma, M.; et al. An intronic variable number of tandem repeat polymorphisms of the cold-induced autoinflammatory syndrome 1 (CIAS1) gene modifies gene expression and is associated with essential hypertension. Eur. J. Hum. Genet. 2006, 14, 1295–1305. [Google Scholar] [CrossRef] [Green Version]
- van den Meiracker, A.H.; Boomsma, F. The angiotensin II-sympathetic nervous system connection. J. Hypertens. 2003, 21, 1453–1454. [Google Scholar] [CrossRef]
- Torretti, J. Sympathetic Control of Renin Release. Annu. Rev. Pharmacol. Toxicol. 1982, 22, 167–192. [Google Scholar] [CrossRef] [PubMed]
- Rust, P.; Ekmekcioglu, C. Impact of Salt Intake on the Pathogenesis and Treatment of Hypertension. Adv. Exp. Med. Biol. 2017, 956, 61–84. [Google Scholar] [CrossRef] [PubMed]
- Platten, M.; Youssef, S.; Hur, E.M.; Ho, P.P.; Han, M.H.; Lanz, T.V.; Phillips, L.K.; Goldstein, M.J.; Bhat, R.; Raine, C.S.; et al. Blocking angiotensin-converting enzyme induces potent regulatory T cells and modulates TH1- and TH17-mediated autoimmunity. Proc. Natl. Acad. Sci. USA 2009, 106, 14948–14953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shirasuna, K.; Karasawa, T.; Usui, F.; Kobayashi, M.; Komada, T.; Kimura, H.; Kawashima, A.; Ohkuchi, A.; Taniguchi, S.; Takahashi, M. NLRP3 Deficiency Improves Angiotensin II-Induced Hypertension But Not Fetal Growth Restriction During Pregnancy. Endocrinology 2015, 156, 4281–4292. [Google Scholar] [CrossRef] [Green Version]
- Kwon, O.C.; Park, Y.; Lee, J.S.; Oh, J.S.; Kim, Y.-G.; Lee, C.-K.; Yoo, B.; Hong, S. Non-albumin proteinuria as a parameter of tubulointerstitial inflammation in lupus nephritis. Clin. Rheumatol. 2019, 38, 235–241. [Google Scholar] [CrossRef]
- Li, X.C.; Zhuo, J.L. Nuclear factor-κB as a hormonal intracellular signaling molecule: Focus on angiotensin II-induced cardiovascular and renal injury. Curr. Opin. Nephrol. Hypertens. 2008, 17, 37–43. [Google Scholar] [CrossRef] [Green Version]
- Grande, M.T.; Pérez-Barriocanal, F.; López-Novoa, J.M. Role of inflammation in túbulo-interstitial damage associated to obstructive nephropathy. J. Inflamm. 2010, 7, 19. [Google Scholar] [CrossRef] [Green Version]
- Tashiro, M.; Sasatomi, Y.; Watanabe, R.; Watanabe, M.; Miyake, K.; Abe, Y.; Yasuno, T.; Ito, K.; Ueki, N.; Hamauchi, A.; et al. IL-1β promotes tubulointerstitial injury in MPO-ANCA-associated glomerulonephritis. Clin. Nephrol. 2016, 86, 190–199. [Google Scholar] [CrossRef]
- Li, Y.; Xia, W.; Wu, M.; Yin, J.; Wang, Q.; Li, S.; Zhang, A.; Huang, S.; Zhang, Y.; Jia, Z. Activation of GSDMD contributes to acute kidney injury induced by cisplatin. Am. J. Physiol.-Ren. Physiol. 2019, 318, F96–F106. [Google Scholar] [CrossRef]
- Neßelhut, T.; Rath, W.; Grospietsch, G.; Weber, M.H.; Kuhn, W. Urinary protein electrophoresis profile in normal and hypertensive pregnancies. Arch. Gynecol. Obstet. 1989, 246, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, T.; Yoshimura, S.; Sasamori, Y.; Sakamoto, T.; Ogino, M.; Kambegawa, A.; Okinaga, S.; Arai, K. Analysis of Urinary Protein by Immunoblot Method Using Unconcentrated Urine in Preeclampsia. Asia. Ocean. J. Obstet. Gynaecol. 1992, 18, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Kaltenbach, F.J.; Boesken, W.H.; Wilhelm, C.; Ziupa, J.; Toussaint, M.N.; Quaas, L. Urinary Protein Patterns and Preeclampsia. Clin. Exp. Hypertens. B 1983, 2, 133–144. [Google Scholar] [CrossRef] [PubMed]
- Engelmann, B.; Massberg, S. Thrombosis as an intravascular effector of innate immunity. Nat. Rev. Immunol. 2013, 13, 34–45. [Google Scholar] [CrossRef] [PubMed]
- Murthy, P.; Durco, F.; Miller-Ocuin, J.L.; Takedai, T.; Shankar, S.; Liang, X.; Liu, X.; Cui, X.; Sachdev, U.; Rath, D.; et al. The NLRP3 inflammasome and bruton’s tyrosine kinase in platelets co-regulate platelet activation, aggregation, and in vitro thrombus formation. Biochem. Biophys. Res. Commun. 2017, 483, 230–236. [Google Scholar] [CrossRef] [PubMed]
- Brown, G.T.; Narayanan, P.; Li, W.; Silverstein, R.L.; McIntyre, T.M. Lipopolysaccharide Stimulates Platelets through an IL-1β Autocrine Loop. J. Immunol. 2013, 191, 5196–5203. [Google Scholar] [CrossRef] [Green Version]
- Brocklebank, V.; Wood, K.M.; Kavanagh, D. Thrombotic Microangiopathy and the Kidney. Clin. J. Am. Soc. Nephrol. 2018, 13, 300–317. [Google Scholar] [CrossRef] [Green Version]
- Vogel, S.; Thein, S.L. Platelets at the crossroads of thrombosis, inflammation and haemolysis. Br. J. Haematol. 2018, 180, 761–767. [Google Scholar] [CrossRef]
- Berhan, Y. Predictors of Perinatal Mortality Associated with Placenta Previa and Placental Abruption: An Experience from a Low Income Country. J. Pregnancy 2014, 2014, 307043. [Google Scholar] [CrossRef] [Green Version]
- Oyelese, Y.; Ananth, C. Placental Abruption. Obstet. Gynecol. 2006, 108, 1005–1016. [Google Scholar] [CrossRef]
- Nath, C.A.; Ananth, C.V.; Smulian, J.C.; Shen-Schwarz, S.; Kaminsky, L. Histologic evidence of inflammation and risk of placental abruption. Am. J. Obstet. Gynecol. 2007, 197, 319.e1–319.e6. [Google Scholar] [CrossRef] [PubMed]
- Brăila, A.D.; Gluhovschi, A.; Neacşu, A.; Lungulescu, C.V.; Brăila, M.; Vîrcan, E.L.; Cotoi, B.V.; Gogănău, A.M. Placental abruption: Etiopathogenic aspects, diagnostic and therapeutic implications. Rom. J. Morphol. Embryol. Rev. Roum. Morphol. Embryol. 2018, 59, 187–195. [Google Scholar]
- Major, H.D.; Campbell, R.A.; Silver, R.M.; Branch, D.W.; Weyrich, A.S. Synthesis of sFlt-1 by platelet-monocyte aggregates contributes to the pathogenesis of preeclampsia. Am. J. Obstet. Gynecol. 2014, 210, 547.e1–547.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karumanchi, S.A.; Maynard, S.E.; Stillman, I.E.; Epstein, F.H.; Sukhatme, V.P. Preeclampsia: A renal perspective. Kidney Int. 2005, 67, 2101–2113. [Google Scholar] [CrossRef] [Green Version]
- Abildgaard, U.; Heimdal, K. Pathogenesis of the syndrome of hemolysis, elevated liver enzymes, and low platelet count (HELLP): A review. Eur. J. Obstet. Gynecol. Reprod. Biol. 2013, 166, 117–123. [Google Scholar] [CrossRef]
- Erdei, J.; Tóth, A.; Balogh, E.; Nyakundi, B.B.; Bányai, E.; Ryffel, B.; Paragh, G.; Cordero, M.D.; Jeney, V. Induction of NLRP3 Inflammasome Activation by Heme in Human Endothelial Cells. Oxid. Med. Cell. Longev. 2018, 2018, 4310816. [Google Scholar] [CrossRef] [Green Version]
- Mustafa, M.F.; Bano, B. Bilirubin binding with liver cystatin induced structural and functional changes. J. Fluoresc. 2014, 24, 967–974. [Google Scholar] [CrossRef] [PubMed]
- Ben-Ari, Z.; Mor, E.; Azarov, D.; Sulkes, J.; Tor, R.; Cheporko, Y.; Hochhauser, E.; Pappo, O. Cathepsin B inactivation attenuates the apoptotic injury induced by ischemia/reperfusion of mouse liver. Apoptosis 2005, 10, 1261–1269. [Google Scholar] [CrossRef] [PubMed]
- Regal, J.F.; Burwick, R.M.; Fleming, S.D. The Complement System and Preeclampsia. Curr. Hypertens. Rep. 2017, 19, 87. [Google Scholar] [CrossRef] [PubMed]
- Burwick Richard, M.; Fichorova Raina, N.; Dawood Hassan, Y.; Yamamoto Hidemi, S.; Feinberg Bruce, B. Urinary Excretion of C5b-9 in Severe Preeclampsia. Hypertension 2013, 62, 1040–1045. [Google Scholar] [CrossRef] [Green Version]
- Chapin, J.; Terry, H.S.; Kleinert, D.; Laurence, J. The role of complement activation in thrombosis and hemolytic anemias. Transfus. Apher. Sci. 2016, 54, 191–198. [Google Scholar] [CrossRef] [PubMed]
- Xu, B.; Jiang, M.; Chu, Y.; Wang, W.; Chen, D.; Li, X.; Zhang, Z.; Zhang, D.; Fan, D.; Nie, Y.; et al. Gasdermin D plays a key role as a pyroptosis executor of non-alcoholic steatohepatitis in humans and mice. J. Hepatol. 2018, 68, 773–782. [Google Scholar] [CrossRef]
- Faletti, L.; Peintner, L.; Neumann, S.; Sandler, S.; Grabinger, T.; Mac Nelly, S.; Merfort, I.; Huang, C.-H.; Tschaharganeh, D.; Kang, T.-W.; et al. TNFα sensitizes hepatocytes to FasL-induced apoptosis by NFκB-mediated Fas upregulation. Cell Death Dis. 2018, 9, 909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stødle, G.S.; Silva, G.B.; Tangerås, L.H.; Gierman, L.M.; Nervik, I.; Dahlberg, U.E.; Sun, C.; Aune, M.H.; Thomsen, L.C.V.; Bjørge, L.; et al. Placental inflammation in preeclampsia by Nod-like receptor protein (NLRP)3 inflammasome activation in trophoblasts. Clin. Exp. Immunol. 2018, 193, 84–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matias, M.L.; Romão, M.; Weel, I.C.; Ribeiro, V.R.; Nunes, P.R.; Borges, V.T.; Araújo, J.P., Jr.; Peraçoli, J.C.; de Oliveira, L.; Peraçoli, M.T. Endogenous and Uric Acid-Induced Activation of NLRP3 Inflammasome in Pregnant Women with Preeclampsia. PLoS ONE 2015, 10, e0129095. [Google Scholar] [CrossRef]
- Nunes, P.R.; Peracoli, M.T.S.; Romao-Veiga, M.; Matias, M.L.; Ribeiro, V.R.; Da Costa Fernandes, C.J.; Peracoli, J.C.; Rodrigues, J.R.; De Oliveira, L. Hydrogen peroxide-mediated oxidative stress induces inflammasome activation in term human placental explants. Pregnancy Hypertens. 2018, 14, 29–36. [Google Scholar] [CrossRef] [Green Version]
- Mulla, M.J.; Myrtolli, K.; Potter, J.; Boeras, C.; Kavathas, P.B.; Sfakianaki, A.K.; Tadesse, S.; Norwitz, E.R.; Guller, S.; Abrahams, V.M. Uric Acid Induces Trophoblast IL-1β Production Via the Inflammasome: Implications for the Pathogenesis of Preeclampsia. Am. J. Reprod. Immunol. 2011, 65, 542–548. [Google Scholar] [CrossRef] [Green Version]
- Romão-Veiga, M.; Matias, M.L.; Ribeiro, V.R.; Nunes, P.R.; M. Borges, V.T.; Peraçoli, J.C.; Peraçoli, M.T.S. Induction of systemic inflammation by hyaluronan and hsp70 in women with preeclampsia. Cytokine 2018, 105, 23–31. [Google Scholar] [CrossRef] [Green Version]
- Han, C.S.; Herrin, M.A.; Pitruzzello, M.C.; Mulla, M.J.; Werner, E.F.; Pettker, C.M.; Flannery, C.A.; Abrahams, V.M. Glucose and Metformin Modulate Human First Trimester Trophoblast Function: A Model and Potential Therapy for Diabetes-Associated Uteroplacental Insufficiency. Am. J. Reprod. Immunol. 2015, 73, 362–371. [Google Scholar] [CrossRef] [Green Version]
- Wiznitzer, A.; Mayer, A.; Novack, V.; Sheiner, E.; Gilutz, H.; Malhotra, A.; Novack, L. Association of lipid levels during gestation with preeclampsia and gestational diabetes mellitus: A population-based study. Am. J. Obstet. Gynecol. 2009, 201, 482.e1–482.e8. [Google Scholar] [CrossRef] [Green Version]
- Yang, F.; Qin, Y.; Wang, Y.; Meng, S.; Xian, H.; Che, H.; Lv, J.; Li, Y.; Yu, Y.; Bai, Y.; et al. Metformin Inhibits the NLRP3 Inflammasome via AMPK/mTOR-dependent Effects in Diabetic Cardiomyopathy. Int. J. Biol. Sci. 2019, 15, 1010–1019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Z.; Zhao, X.; Shan, H.; Gao, H.; Wang, P. microRNA-520c-3p suppresses NLRP3 inflammasome activation and inflammatory cascade in preeclampsia by downregulating NLRP3. Inflamm. Res. 2019, 68, 643–654. [Google Scholar] [CrossRef] [PubMed]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Socha, M.W.; Malinowski, B.; Puk, O.; Dubiel, M.; Wiciński, M. The NLRP3 Inflammasome Role in the Pathogenesis of Pregnancy Induced Hypertension and Preeclampsia. Cells 2020, 9, 1642. https://doi.org/10.3390/cells9071642
Socha MW, Malinowski B, Puk O, Dubiel M, Wiciński M. The NLRP3 Inflammasome Role in the Pathogenesis of Pregnancy Induced Hypertension and Preeclampsia. Cells. 2020; 9(7):1642. https://doi.org/10.3390/cells9071642
Chicago/Turabian StyleSocha, Maciej W., Bartosz Malinowski, Oskar Puk, Mariusz Dubiel, and Michał Wiciński. 2020. "The NLRP3 Inflammasome Role in the Pathogenesis of Pregnancy Induced Hypertension and Preeclampsia" Cells 9, no. 7: 1642. https://doi.org/10.3390/cells9071642