KLF4 Acts as a wt-CFTR Suppressor through an AKT-Mediated Pathway

 , and
, and 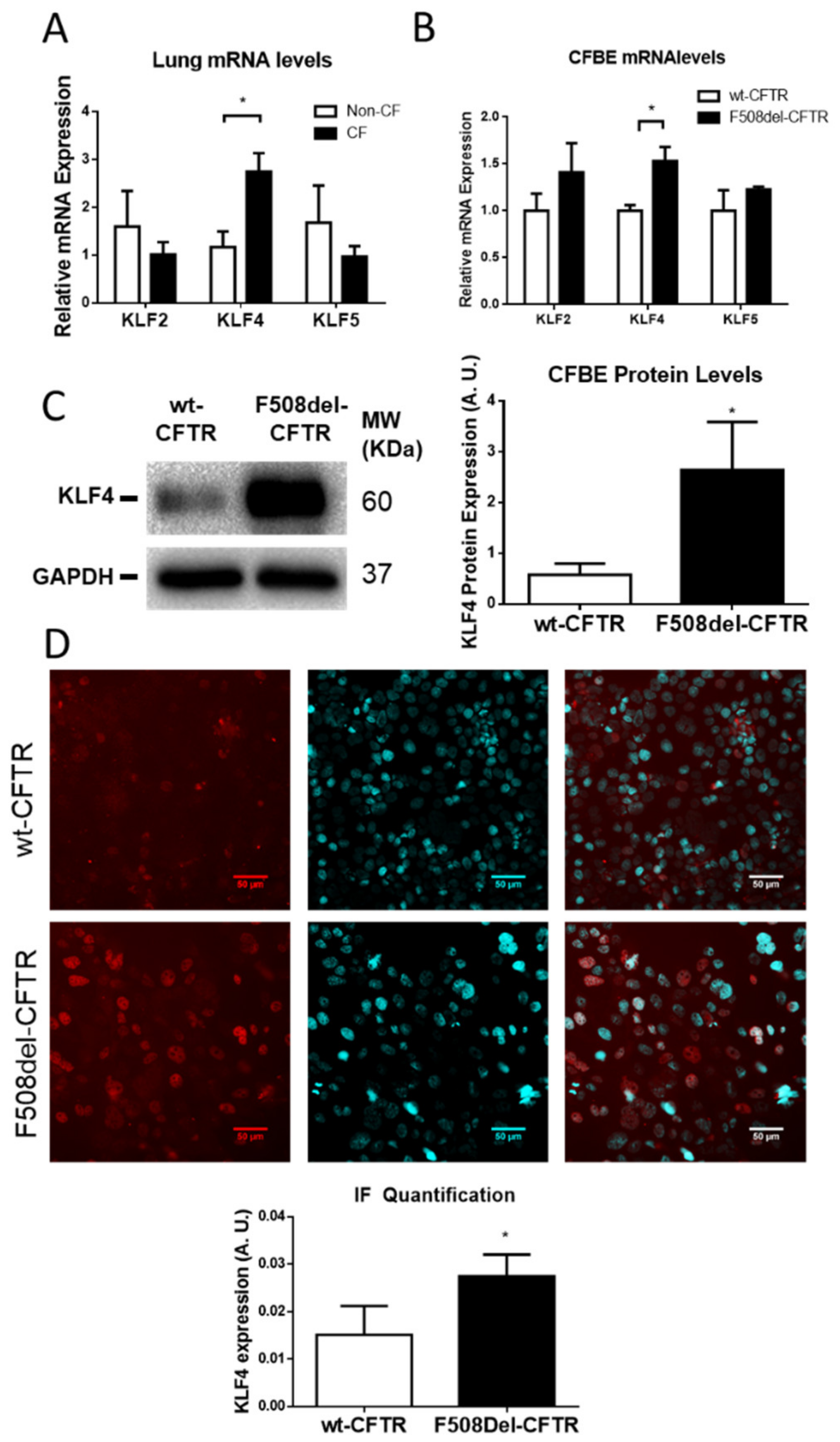{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Human Lungs
2.2. Chemicals, Antibodies, and Primers
2.3. Cell Lines
2.4. KLF4-KO Cells Generation Using CRISPR/Cas9
2.5. RT-qPCR
2.6. Biochemical Assays
2.7. Immunofluorescence Staining (IF)
2.8. Ussing Chamber Experiments
2.9. Patch-Clamp
2.10. Statistical Analyses
3. Results
3.1. KLF4 is Upregulated in CF Native Human Lung and Cell Lines vs. Non-CF
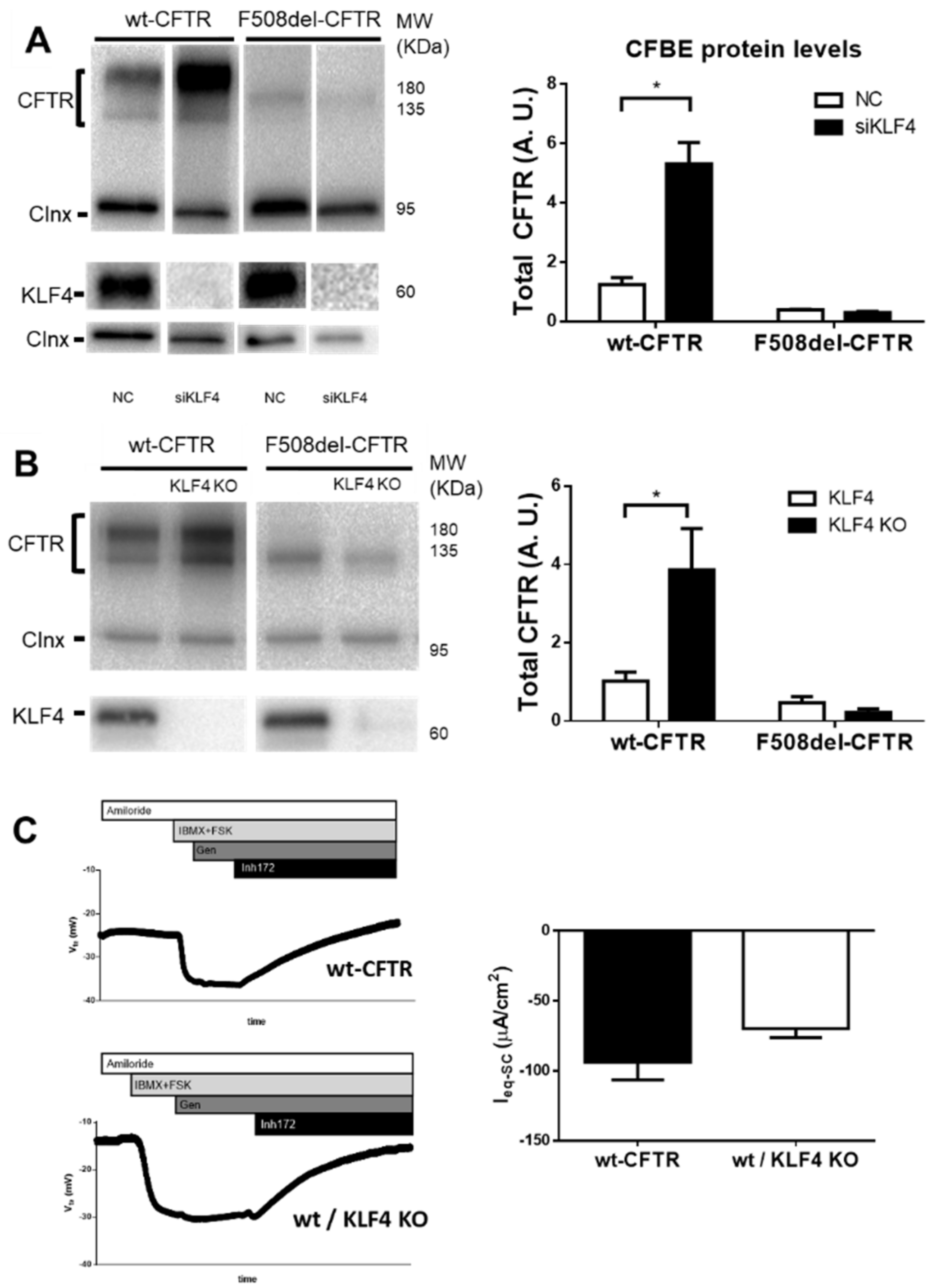
3.2. KLF4 Downregulation Promotes Expression of wt-CFTR But Not of F508del–CFTR
3.3. KLF4 Overexpression Decreases Expression and Function of wt-CFTR
3.4. Characterization of KLF4–CFTR Pathway Crosstalk
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A. Extended Methodology
Appendix A.1. Cell Lines and Growth Conditions
Appendix A.2. Treatment with Compounds
Appendix A.3. siRNA Transfection
Appendix A.4. cDNA Transfection
Appendix A.5. KLF4 KO Cell Generation
Appendix A.6. RNA Extraction, Reverse Transcription, and RT-qPCR
Appendix A.7. Protein Extraction
Appendix A.8. Protein Quantification Assay
Appendix A.9. Western Blot
Appendix A.10. Co-Immunoprecipitation
Appendix A.11. Immunofluorescence Assay
Appendix A.12. Ussing Chamber Experiments
Appendix A.13. Patch-Clamp
Appendix A.14. Statistical Analyses
References
- Zolin, A.; Orenti, A.; Naehrlich, L.; van Rens, J. Ecfspr Annual Report 2017; European Cystic Fibrosis Society: Karup, Denmark, 2017. [Google Scholar]
- Farinha, C.M.; Amaral, M.D. Most F508del-CFTR is targeted to degradation at an early folding checkpoint and independently of calnexin. Mol. Cell. Biol. 2005, 25, 5242–5252. [Google Scholar] [CrossRef] [Green Version]
- Amaral, M.D.; Quaresma, M.C.; Pankonien, I. What Role Does Cftr Play in Development, Differentiation, Regeneration and Cancer? Int. J. Mol. Sci. 2020, 21, 3133. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Wang, X.; Zou, C.; Zhang, J.; Chen, H.; Tsang, L.; Yu, M.K.; Chung, Y.W.; Wang, J.; Dai, Y.; et al. Defective CFTR promotes intestinal proliferation via inhibition of the hedgehog pathway during cystic fibrosis. Cancer Lett. 2019, 446, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Viart, V.; Bergougnoux, A.; Bonini, J.; Varilh, J.; Chiron, R.; Tabary, O.; Molinari, N.; Claustres, M.; Taulan-Cadars, M. Transcription factors and miRNAs that regulate fetal to adult CFTR expression change are new targets for cystic fibrosis. Eur. Respir. J. 2015, 45, 116–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McConnell, B.B.; Yang, V.W. Mammalian Krüppel-like factors in health and diseases. Physiol. Rev. 2010, 90, 1337–1381. [Google Scholar] [CrossRef] [PubMed]
- Lutful Kabir, F.; Ambalavanan, N.; Liu, G.; Li, P.; Solomon, G.M.; Lal, C.V.; Mazur, M.; Halloran, B.; Szul, T.; Gerthoffer, W.T.; et al. MicroRNA-145 Antagonism Reverses TGF-beta Inhibition of F508del CFTR Correction in Airway Epithelia. Am. J. Respir. Crit. Care Med. 2018, 197, 632–643. [Google Scholar] [CrossRef]
- Crespin, S.; Bacchetta, M.; Bou Saab, J.; Tantilipikorn, P.; Bellec, J.; Dudez, T.; Nguyen, T.H.; Kwak, B.R.; Lacroix, J.S.; Huang, S.; et al. Cx26 regulates proliferation of repairing basal airway epithelial cells. Int. J. Biochem. Cell Biol. 2014, 52, 152–160. [Google Scholar] [CrossRef] [Green Version]
- Bischof, J.M.; Ott, C.J.; Leir, S.H.; Gosalia, N.; Song, L.; London, D.; Furey, T.S.; Cotton, C.U.; Crawford, G.E.; Harris, A. A genome-wide analysis of open chromatin in human tracheal epithelial cells reveals novel candidate regulatory elements for lung function. Thorax 2012, 67, 385–391. [Google Scholar] [CrossRef] [Green Version]
- Mutolo, M.J.; Leir, S.H.; Fossum, S.L.; Browne, J.A.; Harris, A. A transcription factor network represses CFTR gene expression in airway epithelial cells. Biochem. J. 2018, 475, 1323–1334. [Google Scholar] [CrossRef]
- Yin, S.; Ray, G.; Kerschner, J.L.; Hao, S.; Perez, A.; Drumm, M.; Browne, J.; Leir, S.H.; Longworth, M.; Harris, A. Functional genomics analysis of human colon organoids identifies key transcription factors. Physiol Genom. 2020, 52, 234–244. [Google Scholar] [CrossRef]
- Rymut, S.M.; Corey, D.A.; Valerio, D.M.; Erokwu, B.O.; Flask, C.A.; Kelley, T.J.; Hodges, C.A. Improved Growth Patterns in Cystic Fibrosis Mice after Loss of Histone Deacetylase 6. Sci. Rep. 2017, 7, 3676. [Google Scholar] [CrossRef] [PubMed]
- Saavedra, M.T.; Patterson, A.D.; West, J.; Randell, S.H.; Riches, D.W.; Malcolm, K.C.; Cool, C.D.; Nick, J.A.; Dinarello, C.A. Abrogation of anti-inflammatory transcription factor LKLF in neutrophil-dominated airways. Am. J. Respir. Cell Mol. Biol. 2008, 38, 679–688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turpaev, K.T. Transcription Factor KLF2 and Its Role in the Regulation of Inflammatory Processes. Biochemistry 2020, 85, 54–67. [Google Scholar] [CrossRef] [PubMed]
- Clarke, L.A.; Sousa, L.; Barreto, C.; Amaral, M.D. Changes in transcriptome of native nasal epithelium expressing F508del-CFTR and intersecting data from comparable studies. Respir. Res. 2013, 14, 38. [Google Scholar] [CrossRef] [Green Version]
- Bebok, Z.; Collawn, J.F.; Wakefield, J.; Parker, W.; Li, Y.; Varga, K.; Sorscher, E.J.; Clancy, J.P. Failure of cAMP agonists to activate rescued deltaF508 CFTR in CFBE41o-airway epithelial monolayers. J. Physiol. 2005, 569, 601–615. [Google Scholar] [CrossRef]
- Carapeto, A.P.; Vitorino, M.V.; Santos, J.D.; Ramalho, S.S.; Robalo, T.; Rodrigues, M.S. Mechanical Properties of Human Bronchial Epithelial Cells Expressing Wt-and Mutant CFTR. Int. J. Mol. Sci. 2020, 21, 2916. [Google Scholar] [CrossRef] [Green Version]
- Mali, P.; Yang, L.; Esvelt, K.M.; Aach, J.; Guell, M.; DiCarlo, J.E.; Norville, J.E.; Church, G.M. RNA-guided human genome engineering via Cas9. Science 2013, 339, 823–826. [Google Scholar] [CrossRef] [Green Version]
- Simões, F.B.; Quaresma, M.C.; Clarke, L.A.; Silva, I.A.; Pankonien, I.; Railean, V.; Kmit, A.; Amaral, M.D. TMEM16A chloride channel does not drive mucus production. Life Sci Alliance 2019, 2, e201900462. [Google Scholar] [CrossRef] [Green Version]
- Farinha, C.M.; Nogueira, P.; Mendes, F.; Penque, D.; Amaral, M.D. The human DnaJ homologue (Hdj)-1/heat-shock protein (Hsp) 40 co-chaperone is required for the in vivo stabilization of the cystic fibrosis transmembrane conductance regulator by Hsp70. Biochem. J. 2002, 366, 797–806. [Google Scholar] [CrossRef] [Green Version]
- Amaral, M.D.; Farinha, C.M.; Matos, P.; Botelho, H.M. Investigating Alternative Transport of Integral Plasma Membrane Proteins from the ER to the Golgi: Lessons from the Cystic Fibrosis Transmembrane Conductance Regulator (CFTR). Methods Mol. Biol. 2016, 1459, 105–126. [Google Scholar] [CrossRef]
- Lérias, J.R.; Pinto, M.C.; Botelho, H.M.; Awatade, N.T.; Quaresma, M.C.; Silva, I.A.L.; Wanitchakool, P.; Schreiber, R.; Pepperkok, R.; Kunzelmann, K.; et al. A novel microscopy-based assay identifies extended synaptotagmin-1 (ESYT1) as a positive regulator of anoctamin 1 traffic. Biochim. Biophys. Acta. Mol. Cell Res. 2018, 1865, 421–431. [Google Scholar] [CrossRef]
- Reilly, R.; Mroz, M.S.; Dempsey, E.; Wynne, K.; Keely, S.J.; McKone, E.F.; Hiebel, C.; Behl, C.; Coppinger, J.A. Targeting the PI3K/Akt/mTOR signalling pathway in Cystic Fibrosis. Sci. Rep. 2017, 7, 7642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vijay, G.V.; Zhao, N.; Den Hollander, P.; Toneff, M.J.; Joseph, R.; Pietila, M.; Taube, J.H.; Sarkar, T.R.; Ramirez-Pena, E.; Werden, S.J.; et al. GSK3β regulates epithelial-mesenchymal transition and cancer stem cell properties in triple-negative breast cancer. Breast Cancer Res. 2019, 21, 37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villarreal, G., Jr.; Zhang, Y.; Larman, H.B.; Gracia-Sancho, J.; Koo, A.; Garcia-Cardena, G. Defining the regulation of KLF4 expression and its downstream transcriptional targets in vascular endothelial cells. Biochem. Biophys. Res. Commun. 2010, 391, 984–989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghaleb, A.M.; Yang, V.W. Kruppel-like factor 4 (KLF4): What we currently know. Gene 2017, 611, 27–37. [Google Scholar] [CrossRef]
- Cui, G.; Stauffer, B.B.; Imhoff, B.R.; Rab, A.; Hong, J.S.; Sorscher, E.J.; McCarty, N.A. VX-770-mediated potentiation of numerous human CFTR disease mutants is influenced by phosphorylation level. Sci. Rep. 2019, 9, 13460. [Google Scholar] [CrossRef] [Green Version]
- Jih, K.Y.; Hwang, T.C. Vx-770 potentiates CFTR function by promoting decoupling between the gating cycle and ATP hydrolysis cycle. Proc. Natl. Acad. Sci. USA 2013, 110, 4404–4409. [Google Scholar] [CrossRef] [Green Version]
- Park, C.S.; Lewis, A.; Chen, T.; Lacorazza, D. Concise Review: Regulation of Self-Renewal in Normal and Malignant Hematopoietic Stem Cells by Krüppel-Like Factor 4. Stem Cells Transl. Med. 2019, 8, 568–574. [Google Scholar] [CrossRef] [Green Version]
- Riordan, J.R. Assembly of functional CFTR chloride channels. Annu. Rev. Physiol. 2005, 67, 701–718. [Google Scholar] [CrossRef]
- Wang, X.; Venable, J.; LaPointe, P.; Hutt, D.M.; Koulov, A.V.; Coppinger, J.; Gurkan, C.; Kellner, W.; Matteson, J.; Plutner, H.; et al. Hsp90 cochaperone Aha1 downregulation rescues misfolding of CFTR in cystic fibrosis. Cell 2006, 127, 803–815. [Google Scholar] [CrossRef] [Green Version]
- Pankow, S.; Bamberger, C.; Calzolari, D.; Martinez-Bartolome, S.; Lavallee-Adam, M.; Balch, W.E.; Yates, J.R., 3rd. F508 CFTR interactome remodelling promotes rescue of cystic fibrosis. Nature 2015, 528, 510–516. [Google Scholar] [CrossRef] [Green Version]
- Lim, S.H.; Legere, E.A.; Snider, J.; Stagljar, I. Recent Progress in CFTR Interactome Mapping and Its Importance for Cystic Fibrosis. Front. Pharmacol. 2017, 8, 997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Canato, S.; Santos, J.D.; Carvalho, A.S.; Aloria, K.; Amaral, M.D.; Matthiesen, R.; Falcao, A.O.; Farinha, C.M. Proteomic interaction profiling reveals KIFC1 as a factor involved in early targeting of F508del-CFTR to degradation. Cell. Mol. Life Sci. 2018, 75, 4495–4509. [Google Scholar] [CrossRef] [PubMed]
- Santos, J.D.; Canato, S.; Carvalho, A.S.; Botelho, H.M. Folding Status Is Determinant over Traffic-Competence in Defining CFTR Interactors in the Endoplasmic Reticulum. Cells 2019, 8, 353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strubberg, A.M.; Liu, J.; Walker, N.M.; Stefanski, C.D.; MacLeod, R.J.; Magness, S.T.; Clarke, L.L. Cftr Modulates Wnt/beta-Catenin Signaling and Stem Cell Proliferation in Murine Intestine. Cell. Mol. Gastroenterol. Hepatol. 2018, 5, 253–271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fishilevich, S.; Nudel, R.; Rappaport, N.; Hadar, R.; Plaschkes, I.; Iny Stein, T.; Rosen, N.; Kohn, A.; Twik, M.; Safran, M.; et al. GeneHancer: Genome-wide integration of enhancers and target genes in GeneCards. Database 2017, 2017, bax028. [Google Scholar] [CrossRef] [Green Version]
- Le Henaff, C.; Mansouri, R.; Modrowski, D.; Zarka, M.; Geoffroy, V.; Marty, C.; Tarantino, N.; Laplantine, E.; Marie, P.J. Increased NF-kappaB and Decreased Wnt-beta-Catenin Signaling Mediate the Reduced Osteoblast Differentiation and Function in F508Delta-CFTR Mice. J. Biol. Chem. 2015, 290, 18009–18017. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.T.; Wang, Y.; Chen, J.J.; Zhang, X.H.; Dong, J.D.; Tsang, L.L.; Huang, X.R.; Cai, Z.; Lan, H.Y.; Jiang, X.H.; et al. Defective CFTR leads to aberrant beta-catenin activation and kidney fibrosis. Sci. Rep. 2017, 7, 5233. [Google Scholar] [CrossRef] [Green Version]
- Liu, K.; Zhang, X.; Zhang, J.T.; Tsang, L.L.; Jiang, X.; Chan, H.C. Defective CFTR- beta-catenin interaction promotes NF-kappaB nuclear translocation and intestinal inflammation in cystic fibrosis. Oncotarget 2016, 7, 64030–64042. [Google Scholar] [CrossRef]
- Castano, Z.; Gordon-Weeks, P.R.; Kypta, R.M. The neuron-specific isoform of glycogen synthase kinase-3beta is required for axon growth. J. Neurochem. 2010, 113, 117–130. [Google Scholar] [CrossRef]
- Trzcinska-Daneluti, A.M.; Chen, A.; Nguyen, L.; Murchie, R.; Jiang, C.; Moffat, J.; Pelletier, L.; Rotin, D. RNA Interference Screen to Identify Kinases That Suppress Rescue of DeltaF508-CFTR. Mol. Cell. Proteom. 2015, 14, 1569–1583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quan, C.; Sun, J.; Lin, Z.; Jin, T.; Dong, B.; Meng, Z.; Piao, J. Ezrin promotes pancreatic cancer cell proliferation and invasion through activating the Akt/mTOR pathway and inducing YAP translocation. Cancer Manag. Res. 2019, 11, 6553–6566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loureiro, C.A.; Matos, A.M.; Dias-Alves, Â.; Pereira, J.F.; Uliyakina, I.; Barros, P.; Amaral, M.D.; Matos, P. A molecular switch in the scaffold NHERF1 enables misfolded CFTR to evade the peripheral quality control checkpoint. Sci. Signal. 2015, 8, ra48. [Google Scholar] [CrossRef] [PubMed]
- Xiong, X.; Schober, M.; Tassone, E.; Khodadadi-Jamayran, A.; Sastre-Perona, A.; Zhou, H.; Tsirigos, A.; Shen, S.; Chang, M.; Melamed, J.; et al. KLF4, A Gene Regulating Prostate Stem Cell Homeostasis, Is a Barrier to Malignant Progression and Predictor of Good Prognosis in Prostate Cancer. Cell Rep. 2018, 25, 3006–3020.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trzcinska-Daneluti, A.M.; Nguyen, L.; Jiang, C.; Fladd, C.; Uehling, D.; Prakesch, M.; Al-awar, R.; Rotin, D. Use of kinase inhibitors to correct ΔF508-CFTR function. Mol. Cell. Proteom. 2012, 11, 745–757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Yu, S.; Wu, Q.; Dou, N.; Li, Y.; Gao, Y. KLF4-Mediated CDH3 Upregulation Suppresses Human Hepatoma Cell Growth and Migration via GSK-3beta Signaling. Int. J. Biol. Sci. 2019, 15, 953–961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cervantes-Camacho, C.; Beltran-Langarica, A.; Ochoa-Uribe, A.K.; Marsch-Moreno, M.; Ayala-Sumuano, J.T.; Velez-delValle, C.; Kuri-Harcuch, W. The transient expression of Klf4 and Klf5 during adipogenesis depends on GSK3beta activity. Adipocyte 2015, 4, 248–255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Than, B.L.; Linnekamp, J.F.; Starr, T.K.; Largaespada, D.A.; Rod, A.; Zhang, Y.; Bruner, V.; Abrahante, J.; Schumann, A.; Luczak, T.; et al. CFTR is a tumor suppressor gene in murine and human intestinal cancer. Oncogene 2016, 35, 4179–4187. [Google Scholar] [CrossRef]
- Faes, S.; Dormond, O. PI3K and AKT: Unfaithful Partners in Cancer. Int. J. Mol. Sci. 2015, 16, 21138–21152. [Google Scholar] [CrossRef] [Green Version]
- Duda, P.; Akula, S.M.; Abrams, S.L.; Steelman, L.S.; Martelli, A.M.; Cocco, L.; Ratti, S. Targeting GSK3 and Associated Signaling Pathways Involved in Cancer. Cells 2020, 9, 1110. [Google Scholar] [CrossRef]
- Slae, M.; Wilschanski, M. Cystic fibrosis: A gastrointestinal cancer syndrome. Lancet. Oncol. 2018, 19, 719–720. [Google Scholar] [CrossRef]
- Yamada, A.; Komaki, Y.; Komaki, F.; Micic, D.; Zullow, S.; Sakuraba, A. Risk of gastrointestinal cancers in patients with cystic fibrosis: A systematic review and meta-analysis. Lancet Oncol. 2018, 19, 758–767. [Google Scholar] [CrossRef]
- Maisonneuve, P.; Marshall, B.C.; Knapp, E.A.; Lowenfels, A.B. Cancer risk in cystic fibrosis: A 20-year nationwide study from the United States. J. Natl. Cancer Inst. 2013, 105, 122–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hajj, R.; Lesimple, P.; Nawrocki-Raby, B.; Birembaut, P.; Puchelle, E.; Coraux, C. Human airway surface epithelial regeneration is delayed and abnormal in cystic fibrosis. J. Pathol. 2007, 211, 340–350. [Google Scholar] [CrossRef]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sousa, L.; Pankonien, I.; Clarke, L.A.; Silva, I.; Kunzelmann, K.; Amaral, M.D. KLF4 Acts as a wt-CFTR Suppressor through an AKT-Mediated Pathway. Cells 2020, 9, 1607. https://doi.org/10.3390/cells9071607
Sousa L, Pankonien I, Clarke LA, Silva I, Kunzelmann K, Amaral MD. KLF4 Acts as a wt-CFTR Suppressor through an AKT-Mediated Pathway. Cells. 2020; 9(7):1607. https://doi.org/10.3390/cells9071607
Chicago/Turabian StyleSousa, Luis, Ines Pankonien, Luka A Clarke, Iris Silva, Karl Kunzelmann, and Margarida D Amaral. 2020. "KLF4 Acts as a wt-CFTR Suppressor through an AKT-Mediated Pathway" Cells 9, no. 7: 1607. https://doi.org/10.3390/cells9071607