Improving Precise CRISPR Genome Editing by Small Molecules: Is there a Magic Potion?


Abstract
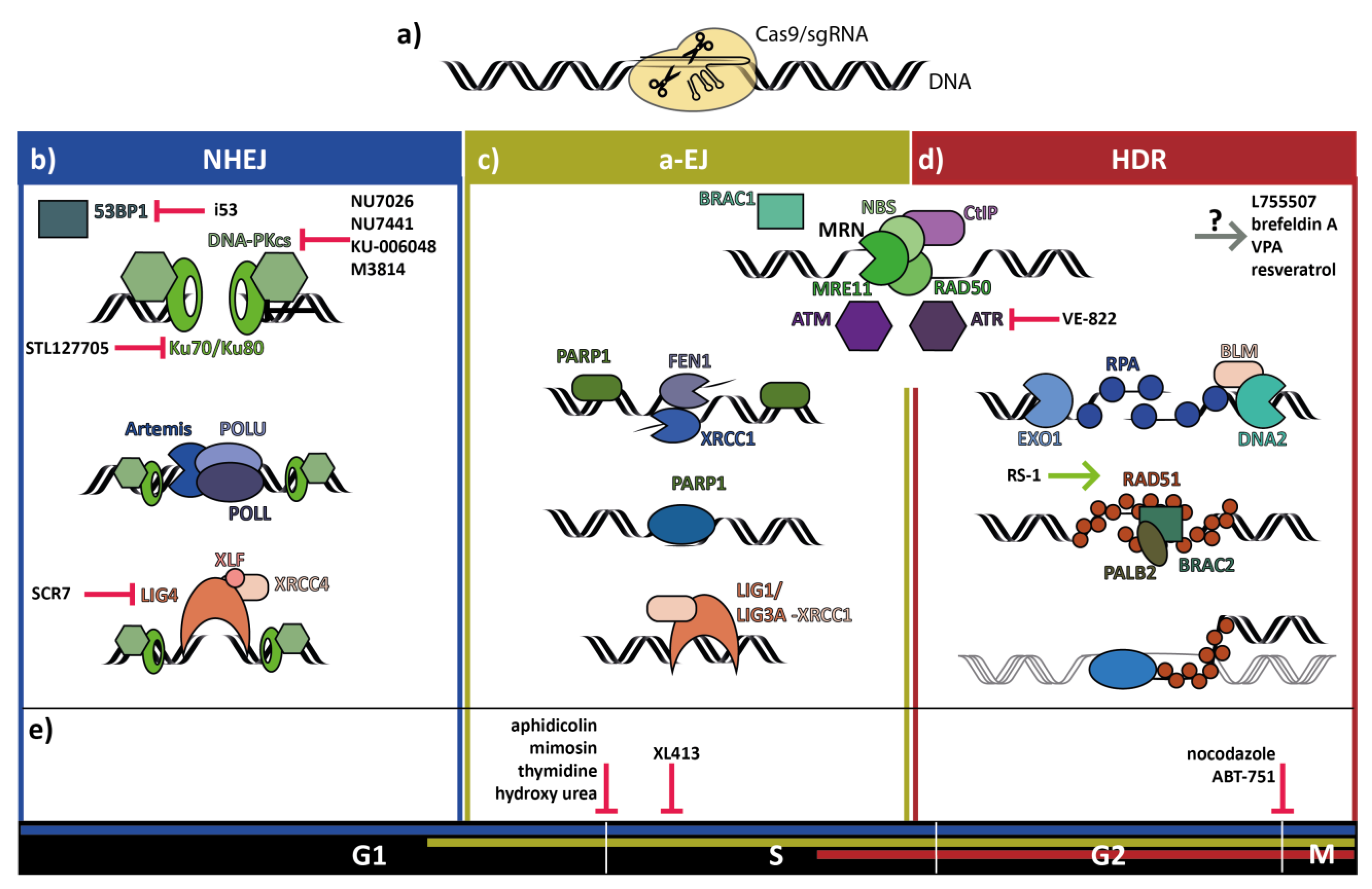
:1. Introduction
2. Non-Homologous End-Joining Repair of Double-Strand Breaks
3. Homology Directed Repair of Double-Strand Breaks
4. Alternative End-Joining Repair of Double-Strand Breaks
5. Double-Strand Break Repair Pathway Choice is Influenced by Cell Cycle Stage
6. Small Molecules to Improve Precise Genome Editing
7. Increase of Homology Directed Repair by Inhibiting Non-Homologous End-Joining
7.1. 53BP1
7.2. Ku70/Ku80
7.3. DNA-PKcs
7.4. DNA Ligase IV
8. Increase of Template-Directed Repair by Facilitation of the Homology Directed Repair Pathway
RAD51
9. Increase of Homology Directed Repair by Cell Cycle Synchronization
10. Increase of Homology Directed Repair Efficiency by Inhibitors via Undetermined Mechanism
11. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Cohen, S.N.; Chang, A.C.Y.; Boyer, H.W.; Helling, R.B. Construction of Biologically Functional Bacterial Plasmids in Vitro. Proc. Natl. Acad. Sci. USA 1973, 70, 3240–3244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Buchholz, F.; Muyrers, J.P.; Stewart, A.F. A new logic for DNA engineering using recombination in Escherichia coli. Nat. Genet. 1998, 20, 123–128. [Google Scholar] [CrossRef] [PubMed]
- Rouet, P.; Smih, F.; Jasin, M. Introduction of double-strand breaks into the genome of mouse cells by expression of a rare-cutting endonuclease. Mol. Cell. Boil. 1994, 14, 8096–8106. [Google Scholar] [CrossRef] [Green Version]
- Maresca, M.; Lin, V.G.; Guo, N.; Yang, Y. Obligate Ligation-Gated Recombination (ObLiGaRe): Custom-designed nuclease-mediated targeted integration through nonhomologous end joining. Genome Res. 2012, 23, 539–546. [Google Scholar] [CrossRef] [Green Version]
- Scully, R.; Panday, A.; Elango, R.; A Willis, N. DNA double-strand break repair-pathway choice in somatic mammalian cells. Nat. Rev. Mol. Cell Boil. 2019, 20, 698–714. [Google Scholar] [CrossRef]
- Walker, J.R.; Corpina, R.A.; Goldberg, J. Structure of the Ku heterodimer bound to DNA and its implications for double-strand break repair. Nature 2001, 412, 607–614. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Crowe, J.L.; Liu, X.; Nakajima, S.; Wang, Y.; Li, C.; Lee, B.J.; Dubois, R.L.; Liu, C.; Yu, X.; et al. Differential phosphorylation of DNA-PKcs regulates the interplay between end-processing and end-ligation during nonhomologous end-joining. Mol. Cell 2015, 58, 172–185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, Y.; Wang, W.; Ding, Q.; Ye, R.; Chen, D.; Merkle, D.; Schriemer, D.; Meek, K.; Lees-Miller, S.P. DNA-PK phosphorylation sites in XRCC4 are not required for survival after radiation or for V(D)J recombination. DNA Repair 2003, 2, 1239–1252. [Google Scholar] [CrossRef]
- Lee, K.-J.; Saha, J.; Sun, J.; Fattah, K.R.; Wang, S.-C.; Jakob, B.; Chi, L.; Wang, S.-Y.; Taucher-Scholz, G.; Davis, A.J.; et al. Phosphorylation of Ku dictates DNA double-strand break (DSB) repair pathway choice in S phase. Nucleic Acids Res. 2015, 44, 1732–1745. [Google Scholar] [CrossRef] [Green Version]
- Uematsu, N.; Weterings, E.; Yano, K.-I.; Morotomi-Yano, K.; Jakob, B.; Taucher-Scholz, G.; Mari, P.-O.; Van Gent, D.C.; Chen, B.P.; Chen, D.J. Autophosphorylation of DNA-PKCS regulates its dynamics at DNA double-strand breaks. J. Cell Boil. 2007, 177, 219–229. [Google Scholar] [CrossRef] [Green Version]
- Ma, Y.; Pannicke, U.; Schwarz, K.; Lieber, M.R. Hairpin Opening and Overhang Processing by an Artemis/DNA-Dependent Protein Kinase Complex in Nonhomologous End Joining and V(D)J Recombination. Cell 2002, 108, 781–794. [Google Scholar] [CrossRef] [Green Version]
- McElhinny, S.A.N.; Havener, J.M.; Garcia-Diaz, M.; Juárez, R.; Bebenek, K.; Kee, B.L.; Blanco, L.; Kunkel, T.; Ramsden, D.A. A Gradient of Template Dependence Defines Distinct Biological Roles for Family X Polymerases in Nonhomologous End Joining. Mol. Cell 2005, 19, 357–366. [Google Scholar] [CrossRef] [PubMed]
- Ahnesorg, P.; Smith, P.; Jackson, S. XLF Interacts with the XRCC4-DNA Ligase IV Complex to Promote DNA Nonhomologous End-Joining. Cell 2006, 124, 301–313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McElhinny, S.A.N.; Snowden, C.M.; McCarville, J.; Ramsden, D.A. Ku Recruits the XRCC4-Ligase IV Complex to DNA Ends. Mol. Cell. Boil. 2000, 20, 2996–3003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guirouilh-Barbat, J.; Huck, S.; Bertrand, P.; Pirzio, L.; Desmaze, C.; Sabatier, L.; Lopez, B.S. Impact of the KU80 Pathway on NHEJ-Induced Genome Rearrangements in Mammalian Cells. Mol. Cell 2004, 14, 611–623. [Google Scholar] [CrossRef] [PubMed]
- Bétermier, M.; Bertrand, P.; Lopez, B.S. Is Non-Homologous End-Joining Really an Inherently Error-Prone Process? PLoS Genet. 2014, 10, e1004086. [Google Scholar] [CrossRef] [Green Version]
- Brinkman, E.; Chen, T.; De Haas, M.; Holland, L.; Akhtar, W.; Van Steensel, B. Kinetics and Fidelity of the Repair of Cas9-Induced Double-Strand DNA Breaks. Mol. Cell 2018, 70, 801–813.e6. [Google Scholar] [CrossRef] [Green Version]
- Sartori, A.; Lukas, C.; Coates, J.; Mistrik, M.; Fu, S.; Bartek, J.; Baer, R.; Lukas, J.; Jackson, S. Human CtIP promotes DNA end resection. Nature 2007, 450, 509–514. [Google Scholar] [CrossRef] [Green Version]
- Maréchal, A.; Zou, L. DNA Damage Sensing by the ATM and ATR Kinases. Cold Spring Harb. Perspect. Boil. 2013, 5, a012716. [Google Scholar] [CrossRef]
- Nimonkar, A.V. BLM-DNA2-RPA-MRN and EXO1-BLM-RPA-MRN constitute two DNA end resection machineries for human DNA break repair. Genes Dev. 2011, 25, 350–362. [Google Scholar] [CrossRef] [Green Version]
- Whelan, D.R.; Lee, W.T.C.; Yin, Y.; Ofri, D.M.; Bermudez-Hernandez, K.; Keegan, S.; Fenyo, D.; Rothenberg, E. Spatiotemporal dynamics of homologous recombination repair at single collapsed replication forks. Nat. Commun. 2018, 9, 3882. [Google Scholar] [CrossRef] [Green Version]
- Danilowicz, C.; Peacock-Villada, A.; Vlassakis, J.; Facon, A.; Feinstein, E.; Kleckner, N.; Prentiss, M. The differential extension in dsDNA bound to Rad51 filaments may play important roles in homology recognition and strand exchange. Nucleic Acids Res. 2013, 42, 526–533. [Google Scholar] [CrossRef] [Green Version]
- Wright, W.D.; Shah, S.S.; Heyer, W.-D. Homologous recombination and the repair of DNA double-strand breaks. J. Boil. Chem. 2018, 293, 10524–10535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Truong, L.N.; Li, Y.; Shi, L.Z.; He, J.; Razavian, N.; Berns, M.W.; Wu, X.; Hwang, P.Y.-H.; Wang, H. Microhomology-mediated End Joining and Homologous Recombination share the initial end resection step to repair DNA double-strand breaks in mammalian cells. Proc. Natl. Acad. Sci. USA 2013, 110, 7720–7725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caron, M.-C.; Sharma, A.K.; O’Sullivan, J.; Myler, L.R.; Ferreira, M.T.; Rodrigue, A.; Coulombe, Y.; Ethier, C.; Gagné, J.-P.; Langelier, M.-F.; et al. Poly(ADP-ribose) polymerase-1 antagonizes DNA resection at double-strand breaks. Nat. Commun. 2019, 10, 2954. [Google Scholar] [CrossRef] [Green Version]
- Sharma, S.; Javadekar, S.M.; Pandey, M.; Srivastava, M.; Kumari, R.; Raghavan, S.C. Homology and enzymatic requirements of microhomology-dependent alternative end joining. Cell Death Dis. 2015, 6, e1697. [Google Scholar] [CrossRef] [Green Version]
- Kent, T. Mechanism of microhomology-mediated end-joining promoted by human DNA polymerase theta. Nat. Struct. Mol. Biol. 2015, 22, 230–237. [Google Scholar] [CrossRef] [Green Version]
- Lü, G.; Duan, J.; Shu, S.; Wang, X.; Gao, L.; Guo, J.; Zhang, Y. Ligase I and ligase III mediate the DNA double-strand break ligation in alternative end-joining. Proc. Natl. Acad. Sci. USA 2016, 113, 1256–1260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sallmyr, A.; E Tomkinson, A. Repair of DNA double-strand breaks by mammalian alternative end-joining pathways. J. Boil. Chem. 2018, 293, 10536–10546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orthwein, A.; Noordermeer, S.; Wilson, M.D.; Landry, S.; Enchev, R.I.; Sherker, A.; Munro, M.; Pinder, J.; Salsman, J.; Dellaire, G.; et al. A mechanism for the suppression of homologous recombination in G1 cells. Nature 2015, 528, 422–426. [Google Scholar] [CrossRef]
- Isono, M.; Niimi, A.; Oike, T.; Hagiwara, Y.; Sato, H.; Sekine, R.; Yoshida, Y.; Isobe, S.-Y.; Obuse, C.; Nishi, R.; et al. BRCA1 Directs the Repair Pathway to Homologous Recombination by Promoting 53BP1 Dephosphorylation. Cell Rep. 2017, 18, 520–532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beumer, K.J.; Trautman, J.K.; Bozas, A.; Liu, J.-L.; Rutter, J.; Gall, J.G.; Carroll, D. Efficient gene targeting in Drosophila by direct embryo injection with zinc-finger nucleases. Proc. Natl. Acad. Sci. USA 2008, 105, 19821–19826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riesenberg, S.; Chintalapati, M.; Macak, D.; Kanis, P.; Maricic, T.; Pääbo, S. Simultaneous precise editing of multiple genes in human cells. Nucleic Acids Res. 2019, 47, e116. [Google Scholar] [CrossRef]
- Shy, B.R.; MacDougall, M.; Clarke, R.; Merrill, B.J. Co-incident insertion enables high efficiency genome engineering in mouse embryonic stem cells. Nucleic Acids Res. 2016, 44, 7997–8010. [Google Scholar] [CrossRef] [PubMed]
- Chu, V.T.; Weber, T.; Wefers, B.; Wurst, W.; Sander, S.; Rajewsky, K.; Kühn, R. Increasing the efficiency of homology-directed repair for CRISPR-Cas9-induced precise gene editing in mammalian cells. Nat. Biotechnol. 2015, 33, 543–548. [Google Scholar] [CrossRef] [Green Version]
- Vispé, S.; Cazaux, C.; Lesca, C.; Defais, M. Overexpression of Rad51 protein stimulates homologous recombination and increases resistance of mammalian cells to ionizing radiation. Nucleic Acids Res. 1998, 26, 2859–2864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tran, N.-T.; Bashir, S.; Li, X.; Rossius, J.; Chu, V.T.; Rajewsky, K.; Kühn, R. Enhancement of Precise Gene Editing by the Association of Cas9 With Homologous Recombination Factors. Front. Genet. 2019, 10, 365. [Google Scholar] [CrossRef] [Green Version]
- Kawasumi, M.; Nghiem, P. Chemical Genetics: Elucidating Biological Systems with Small-Molecule Compounds. J. Investig. Dermatol. 2007, 127, 1577–1584. [Google Scholar] [CrossRef]
- Weiss, W.A.; Taylor, S.S.; Shokat, K.M. Recognizing and exploiting differences between RNAi and small-molecule inhibitors. Nat. Methods 2007, 3, 739–744. [Google Scholar] [CrossRef]
- Richardson, C.D.; Kazane, K.; Feng, S.J.; Zelin, E.; Bray, N.L.; Schäfer, A.J.; Floor, S.N.; Corn, J.E. CRISPR–Cas9 genome editing in human cells occurs via the Fanconi anemia pathway. Nat. Genet. 2018, 50, 1132–1139. [Google Scholar] [CrossRef]
- Wienert, B.; Nguyen, D.N.; Guenther, A.; Feng, S.J.; Locke, M.N.; Wyman, S.K.; Shin, J.; Kazane, K.R.; Gregory, G.L.; Carter, M.A.M.; et al. Timed inhibition of CDC7 increases Crispr-Cas9 mediated templated repair. Nat. Commun. 2020, 11, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Canny, M.D.; Moatti, N.; Wan, L.C.; Fradet-Turcotte, A.; Krasner, D.; Mateos-Gómez, P.A.; Zimmermann, M.; Orthwein, A.; Juang, Y.-C.; Zhang, W.; et al. Inhibition of 53BP1 favors homology-dependent DNA repair and increases CRISPR-Cas9 genome-editing efficiency. Nat. Biotechnol. 2017, 36, 95–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weterings, E. A novel small molecule inhibitor of the DNA repair protein Ku70/80. DNA Repair 2016, 43, 98–106. [Google Scholar] [CrossRef] [PubMed]
- Robert, F.; Barbeau, M.; Ethier, S.; Dostie, J.; Pelletier, J. Pharmacological inhibition of DNA-PK stimulates Cas9-mediated genome editing. Genome Med. 2015, 7, 93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.-P.; Li, X.-L.; Li, G.-H.; Chen, W.; Arakaki, C.; Botimer, G.D.; Baylink, D.; Zhang, L.; Wen, W.; Fu, Y.-W.; et al. Efficient precise knockin with a double cut HDR donor after CRISPR/Cas9-mediated double-stranded DNA cleavage. Genome Boil. 2017, 18, 35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riesenberg, S.; Maricic, T. Targeting repair pathways with small molecules increases precise genome editing in pluripotent stem cells. Nat. Commun. 2018, 9, 2164. [Google Scholar] [CrossRef] [Green Version]
- Yao, X.; Wang, X.; Hu, X.; Liu, Z.; Liu, J.; Zhou, H.; Shen, X.; Wei, Y.; Huang, Z.; Ying, W.; et al. Homology-mediated end joining-based targeted integration using Crispr/Cas9. Cell Res. 2017, 27, 801–814. [Google Scholar] [CrossRef] [Green Version]
- Yao, X.; Zhang, M.; Wang, X.; Ying, W.; Hu, X.; Dai, P.; Meng, F.-L.; Shi, L.; Sun, Y.; Yao, N.; et al. Tild-Crispr Allows for Efficient and Precise Gene Knockin in Mouse and Human Cells. Dev. Cell 2018, 45, 526–536.e5. [Google Scholar] [CrossRef] [Green Version]
- Ma, X.; Chen, X.; Jin, Y.; Ge, W.; Wang, W.; Kong, L.; Ji, J.; Guo, X.; Huang, J.; Feng, X.-H.; et al. Small molecules promote Crispr-Cpf1-mediated genome editing in human pluripotent stem cells. Nat. Commun. 2018, 9, 1303. [Google Scholar] [CrossRef] [Green Version]
- Srivastava, M.; Nambiar, I.; Sharma, S.; Karki, S.S.; Goldsmith, G.; Hegde, M.; Kumar, S.; Pandey, M.; Singh, R.K.; Ray, P.; et al. An Inhibitor of Nonhomologous End-Joining Abrogates Double-Strand Break Repair and Impedes Cancer Progression. Cell 2012, 151, 1474–1487. [Google Scholar] [CrossRef] [Green Version]
- Maruyama, T.; Dougan, S.K.; Truttmann, M.; Bilate, A.M.; Ingram, J.R.; Ploegh, H.L. Increasing the efficiency of precise genome editing with Crispr-Cas9 by inhibition of nonhomologous end joining. Nat. Biotechnol. 2015, 33, 538–542. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.; Schimenti, J.C.; Bolcun-Filas, E. A mouse geneticist’s practical guide to CRISPR applications. Genetics 2014, 199, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, Z.; Shi, Z.; Guo, X.; Jiang, B.; Wang, G.; Luo, D.; Chen, Y.; Zhu, Y.-S. Ligase IV inhibitor SCR7 enhances gene editing directed by Crispr–Cas9 and ssODN in human cancer cells. Cell Biosci. 2018, 8, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, Y.; Chen, W.; Zhang, X.; Yu, L.; Dong, W.; Pan, S.; Gao, S.; Huang, X.; Zhang, L. Increasing the efficiency of Crispr/Cas9-mediated precise genome editing in rats by inhibiting NHEJ and using Cas9 protein. RNA Boil. 2016, 13, 605–612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, G.; Zhang, X.; Zhong, C.; Mo, J.; Quan, R.; Yang, J.; Liu, D.; Li, Z.; Yang, H.; Wu, Z. Small molecules enhance Crispr/Cas9-mediated homology-directed genome editing in primary cells. Sci. Rep. 2017, 7, 8943. [Google Scholar] [CrossRef]
- Gerlach, M.; Kraft, T.; Brenner, B.; Petersen, B.; Niemann, H.; Montag, J. Efficient Knock-in of a Point Mutation in Porcine Fibroblasts Using the Crispr/Cas9-GMNN Fusion Gene. Genes 2018, 9, 296. [Google Scholar] [CrossRef] [Green Version]
- Yang, D. Enrichment of G2/M cell cycle phase in human pluripotent stem cells enhances HDR-mediated gene repair with customizable endonucleases. Sci. Rep. 2016, 6, 21264. [Google Scholar] [CrossRef] [Green Version]
- Song, J.; Yang, D.; Xu, J.; Zhu, T.; Chen, Y.E.; Zhang, J. RS-1 enhances Crispr/Cas9 and TALEN-mediated knock-in efficiency. Nat. Commun. 2016, 7, 10548. [Google Scholar] [CrossRef] [Green Version]
- Jayathilaka, K.; Sheridan, S.D.; Bold, T.D.; Bochenska, K.; Logan, H.L.; Weichselbaum, R.R.; Bishop, U.K.; Connell, P.P. A chemical compound that stimulates the human homologous recombination protein RAD51. Proc. Natl. Acad. Sci. USA 2008, 105, 15848–15853. [Google Scholar] [CrossRef] [Green Version]
- Pinder, J.; Salsman, J.; Dellaire, G. Nuclear domain ’knock-in’ screen for the evaluation and identification of small molecule enhancers of CRISPR-based genome editing. Nucleic Acids Res. 2015, 43, 9379–9392. [Google Scholar] [CrossRef] [Green Version]
- Lamas-Toranzo, I. RS-1 enhances CRISPR-mediated targeted knock-in in bovine embryost. Mol. Reprod. Dev. 2020. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Staahl, B.T.; Alla, R.K.; Doudna, J.A. Enhanced homology-directed human genome engineering by controlled timing of CRISPR/Cas9 delivery. Life 2014, 3, 04766. [Google Scholar] [CrossRef]
- Yu, C.; Liu, Y.; Ma, T.; Liu, K.; Xu, S.; Zhang, Y.; Liu, H.; La Russa, M.; Xie, M.; Ding, S.; et al. Small molecules enhance CRISPR genome editing in pluripotent stem cells. Cell Stem Cell 2015, 16, 142–147. [Google Scholar] [CrossRef] [Green Version]
- Takayama, K.; Igai, K.; Hagihara, Y.; Hashimoto, R.; Hanawa, M.; Sakuma, T.; Tachibana, M.; Sakurai, F.; Yamamoto, T.; Mizuguchi, H. Highly efficient biallelic genome editing of human ES/iPS cells using a Crispr/Cas9 or TALEN system. Nucleic Acids Res. 2017, 45, 5198–5207. [Google Scholar] [CrossRef] [PubMed]
- Fradet-Turcotte, A.; Canny, M.D.; Escribano-Diaz, C.; Orthwein, A.; Leung, C.C.Y.; Huang, H.; Landry, M.-C.; Kitevski-LeBlanc, J.; Noordermeer, S.; Sicheri, F.; et al. 53BP1 is a reader of the DNA-damage-induced H2A Lys 15 ubiquitin mark. Nature 2013, 499, 50–54. [Google Scholar] [CrossRef] [Green Version]
- Paulsen, B.S.; Mandal, P.K.; Frock, R.L.; Boyraz, B.; Yadav, R.; Upadhyayula, S.; Gutierrez-Martinez, P.; Ebina, W.; Fasth, A.; Kirchhausen, T.; et al. Ectopic expression of RAD52 and dn53BP1 improves homology-directed repair during CRISPR-Cas9 genome editing. Nat. Biomed. Eng. 2017, 1, 878–888. [Google Scholar] [CrossRef]
- Jayavaradhan, R.; Pillis, D.M.; Goodman, M.; Zhang, F.; Zhang, Y.; Andreassen, P.R.; Malik, P. Crispr-Cas9 fusion to dominant-negative 53BP1 enhances HDR and inhibits NHEJ specifically at Cas9 target sites. Nat. Commun. 2019, 10, 2866. [Google Scholar] [CrossRef] [Green Version]
- Mohiuddin, I.S.; Kang, M.H. DNA-PK as an Emerging Therapeutic Target in Cancer. Front. Oncol. 2019, 9, 635. [Google Scholar] [CrossRef]
- Beumer, K.J.; Trautman, J.K.; Mukherjee, K.; Carroll, D. Donor DNA Utilization during Gene Targeting with Zinc-Finger Nucleases. G3 Genes Genomes Genetics 2013, 3, 657–664. [Google Scholar] [CrossRef] [Green Version]
- Frank, K.M.; Sekiguchi, J.; Seidl, K.J.; Swat, W.; Rathbun, G.A.; Cheng, H.-L.; Davidson, L.; Kangaloo, L.; Alt, F.W. Late embryonic lethality and impaired V (D)J recombination in mice lacking DNA ligase IV. Nature 1998, 396, 173–177. [Google Scholar] [CrossRef]
- Takata, M. The Rad51 paralog Rad51B promotes homologous recombinational repair. Mol. Cell. Biol. 2000, 20, 6476–6482. [Google Scholar] [CrossRef]
- Takata, M.; Sasaki, M.S.; Tachiiri, S.; Fukushima, T.; Sonoda, E.; Schild, D.; Thompson, L.H.; Takeda, S. Chromosome Instability and Defective Recombinational Repair in Knockout Mutants of the Five Rad51 Paralogs. Mol. Cell. Boil. 2001, 21, 2858–2866. [Google Scholar] [CrossRef] [Green Version]
- Heyer, W.-D.; Ehmsen, K.T.; Liu, J. Regulation of homologous recombination in eukaryotes. Annu. Rev. Genet. 2010, 44, 113–139. [Google Scholar] [CrossRef] [Green Version]
- Koltun, E.S.; Tsuhako, A.L.; Brown, D.S.; Aay, N.; Arcalas, A.; Chan, V.; Du, H.; Engst, S.; Ferguson, K.; Franzini, M.; et al. Discovery of XL413, a potent and selective CDC7 inhibitor. Bioorganic Med. Chem. Lett. 2012, 22, 3727–3731. [Google Scholar] [CrossRef]
- Gutschner, T.; Haemmerle, M.; Genovese, G.; Draetta, G.; Chin, L. Post-translational Regulation of Cas9 during G1 Enhances Homology-Directed Repair. Cell Rep. 2016, 14, 1555–1566. [Google Scholar] [CrossRef] [Green Version]
- Defoort, E.N.; Kim, P.M.; Winn, L.M. Valproic Acid Increases Conservative Homologous Recombination Frequency and Reactive Oxygen Species Formation: A Potential Mechanism for Valproic Acid-Induced Neural Tube Defects. Mol. Pharmacol. 2005, 69, 1304–1310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, Y.; Foden, J.A.; Khayter, C.; Maeder, M.L.; Reyon, D.; Joung, J.K.; Sander, J.D. High-frequency off-target mutagenesis induced by Crispr-Cas nucleases in human cells. Nat. Biotechnol. 2013, 31, 822–826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kosicki, M.; Tomberg, K.; Bradley, A. Repair of double-strand breaks induced by Crispr–Cas9 leads to large deletions and complex rearrangements. Nat. Biotechnol. 2018, 36, 765–771. [Google Scholar] [CrossRef]
- Janssen, J.M.; Chen, X.; Liu, J.; Gonçalves, M.A.F.V. The Chromatin Structure of Crispr-Cas9 Target DNA Controls the Balance between Mutagenic and Homology-Directed Gene-Editing Events. Mol. Ther. Nucleic Acids 2019, 16, 141–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, X.; Gao, H.; Yang, Y.; He, M.; Wu, Y.; Song, Y.; Tong, Y.; Rao, Y. Protacs: Great opportunities for academia and industry. Signal Transduct. Target. Ther. 2019, 4, 64–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, K.; Tsunekawa, Y.; Hernández-Benítez, R.; Wu, J.; Zhu, J.; Kim, E.J.; Hatanaka, F.; Yamamoto, M.; Araoka, T.; Li, Z.; et al. In vivo genome editing via Crispr/Cas9 mediated homology-independent targeted integration. Nature 2016, 540, 144–149. [Google Scholar] [CrossRef] [PubMed]
- Sakuma, T.; Nakade, S.; Sakane, Y.; Suzuki, K.-I.; Yamamoto, T. MMEJ-assisted gene knock-in using TALENs and Cripr-Cas9 with the PITCh systems. Nat. Protoc. 2015, 11, 118–133. [Google Scholar] [CrossRef] [PubMed]
- Komor, A.C.; Badran, A.H.; Liu, D.R. Editing the Genome without Double-Stranded DNA Breaks. ACS Chem. Boil. 2017, 13, 383–388. [Google Scholar] [CrossRef] [PubMed]
- Anzalone, A.V.; Randolph, P.B.; Davis, J.R.; Sousa, A.A.; Koblan, L.W.; Levy, J.M.; Chen, P.J.; Wilson, C.; Newby, G.A.; Raguram, A.; et al. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature 2019, 576, 149–157. [Google Scholar] [CrossRef] [PubMed]
- Klompe, S.E.; Vo, P.L.H.; Halpin-Healy, T.S.; Sternberg, S.H. Transposon-encoded Crispr-Cas systems direct RNA-guided DNA integration. Nature 2019, 571, 219–225. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Small Molecule | Target | Observed Effects | Sources |
|---|---|---|---|
| i53 | Prevents interaction of 53BP1 with ubiquitylated histones at DSBs | Increased HDR with ss and ds donor in several cell lines | [42] |
| STL127705 | Inhibits interaction of Ku proteins with DNA and Ku-dependent PKcs activation | Not tested | [43] |
| NU7441 | Inhibition of DNA-PKcs | Reduces NHEJ and increases HDR in HEK293T cells using ss or ds donors Minor increase in hiPSC with ds donor | [44,45] |
| KU-0060648 | Inhibition of DNA-PKcs | Reduces NHEJ and increases HDR in HEK293T cells using ss or ds donors | [44] |
| NU7026 | Inhibition of DNA-PKcs | Increased KI with ss donor after Cas9 induced DSB or double nicking or Cpf1 induced DSB in hiPSC No effect in mouse embryonic stem cells or in mouse zygotes with ds donor | [34,46,47,48] |
| M3814 | Inhibition of DNA-PKcs | Increased KI in hiPSC and K562 cells with ss donor using Cas9 or Cpf1 | [33] |
| VE-822 | Inhibition of ATR | Increases HDR in hiPSC with ss or ds donor in combination with Cpf1 | [49] |
| SCR7 | Inhibitor of Ligase IV | Decreased NHEJ repair of an extrachromosomal reporter system in HeLa cells Increased HDR in several cell lines with ds donor or ss donor Increased HDR in mice with ss donor No HDR increase in rabbits with ds donor Increased HDR in rats with ds donor No HDR increase for an extrachromosomal reporter in H1 cells Inconsistent effects on HDR efficiency in fetal porcine fibroblasts | [35,50,51,52,53,54,55,56,57,58] |
| RS-1 | Enhances RAD51 binding to ssDNA after end-resection | Increased HDR in cell lines and rabbit embryos with ds donor Increased HDR in bovine embryos with ss donor | [58,59,60,61] |
| Aphidicolin | G1/S blocker | HDR promoting effect in HEK293T and neonatal fibroblasts with ss donor | [62] |
| Mimosin, thymidine, hydroxy urea | G1/S blocker | Increased HDR in neonatal fibroblasts with ss donor Decreased HDR in HEK293T cells with ss donor | [62] |
| Nocodazole | G2/M blocker | HDR promoting effect in HEK293T cells with ss or ds donor No HDR increase in neonatal fibroblasts or human ESC using ss donor Increased HDR in hPSC with ds donor | [57,62] |
| ABT-751 | G2/M blocker | Increased HDR in hPSC with ds donor | [57] |
| XL413 | G1/S blocker | Increased HDR in K562 cells and T cells with ss or ds donor Increase in HSPCs with ss donor (ds not tested) Additionally tested cell lines showed either varying or no effects | [41] |
| L755507 | β3-adrenergic receptor agonist | Increase in HDR in several cell lines with ss and ds donors | [55,63] |
| Brefeldin A | Inhibition of intracellular transport from ER to Golgi | Increase in HDR in mES cells with ds donor | [63] |
| Resveratrol | Broad range of biological activities | Increase of HDR in porcine fetal fibroblasts with ds donor | [55] |
| VPA | HDAC inhibitor | Increase of HDR in human ESC with ds donor | [64] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bischoff, N.; Wimberger, S.; Maresca, M.; Brakebusch, C. Improving Precise CRISPR Genome Editing by Small Molecules: Is there a Magic Potion? Cells 2020, 9, 1318. https://doi.org/10.3390/cells9051318
Bischoff N, Wimberger S, Maresca M, Brakebusch C. Improving Precise CRISPR Genome Editing by Small Molecules: Is there a Magic Potion? Cells. 2020; 9(5):1318. https://doi.org/10.3390/cells9051318
Chicago/Turabian StyleBischoff, Nadja, Sandra Wimberger, Marcello Maresca, and Cord Brakebusch. 2020. "Improving Precise CRISPR Genome Editing by Small Molecules: Is there a Magic Potion?" Cells 9, no. 5: 1318. https://doi.org/10.3390/cells9051318