Pipeline for the Generation and Characterization of Transgenic Human Pluripotent Stem Cells Using the CRISPR/Cas9 Technology

 , and
, and 
Abstract
:1. Introduction
1.1. The CRISPR/Cas9 System
1.2. DNA Repair Mechanisms
2. Preparing the Experiment
2.1. Defining the Project Goal
2.2. Defining the Mutagenesis Event to Be Generated
2.3. Selecting Reagents and Transfection Strategies
2.4. Characterizing the Targeted Sequence
2.5. Anticipating the Screening Strategy
2.6. sgRNA Design and Selection
2.7. DNA Donor Template Design
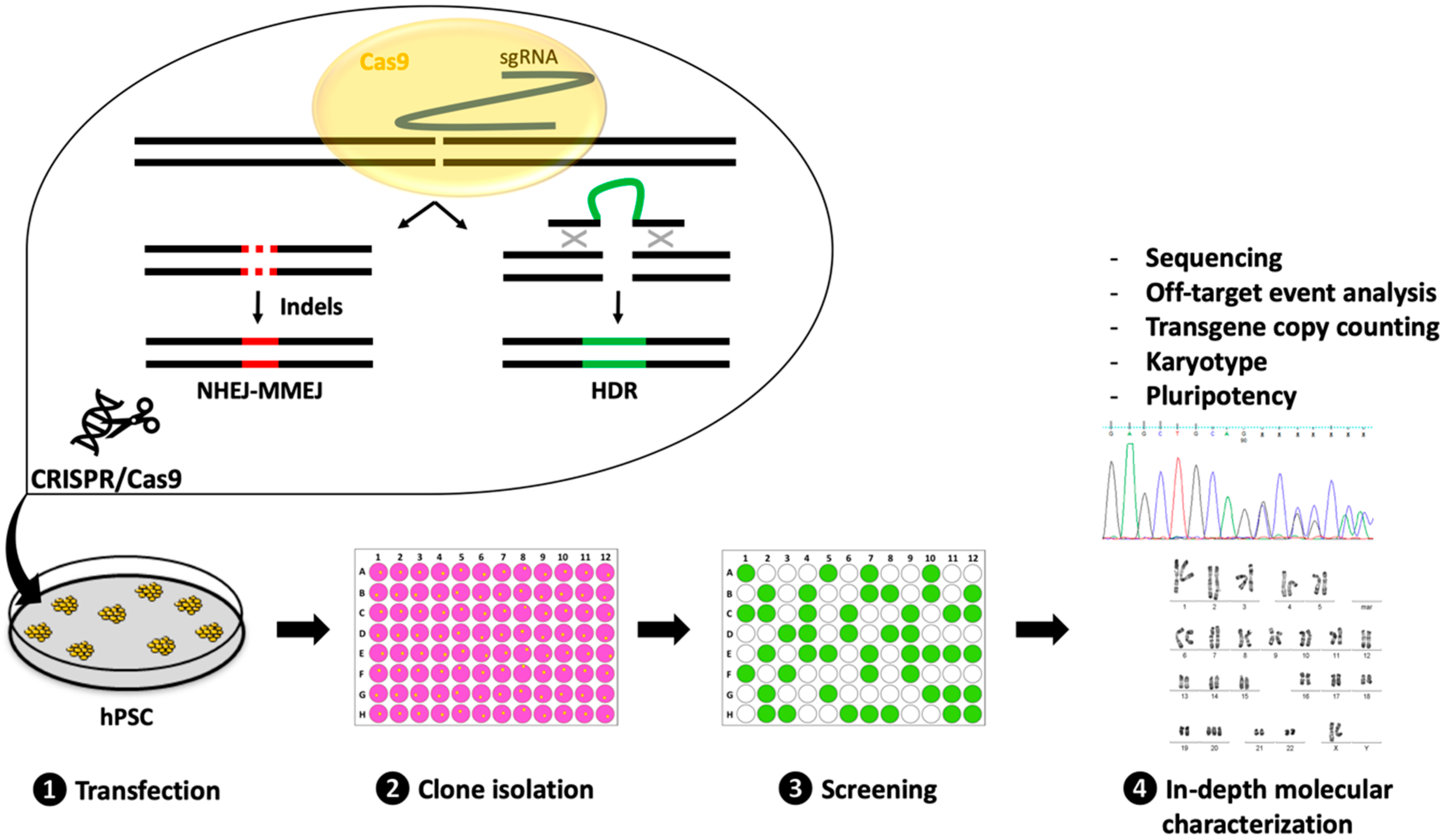
3. Experimental Strategies for the Successful Generation and Characterization of CRISPR-Edited hPSC Lines
3.1. Transfection
3.2. Clone Isolation
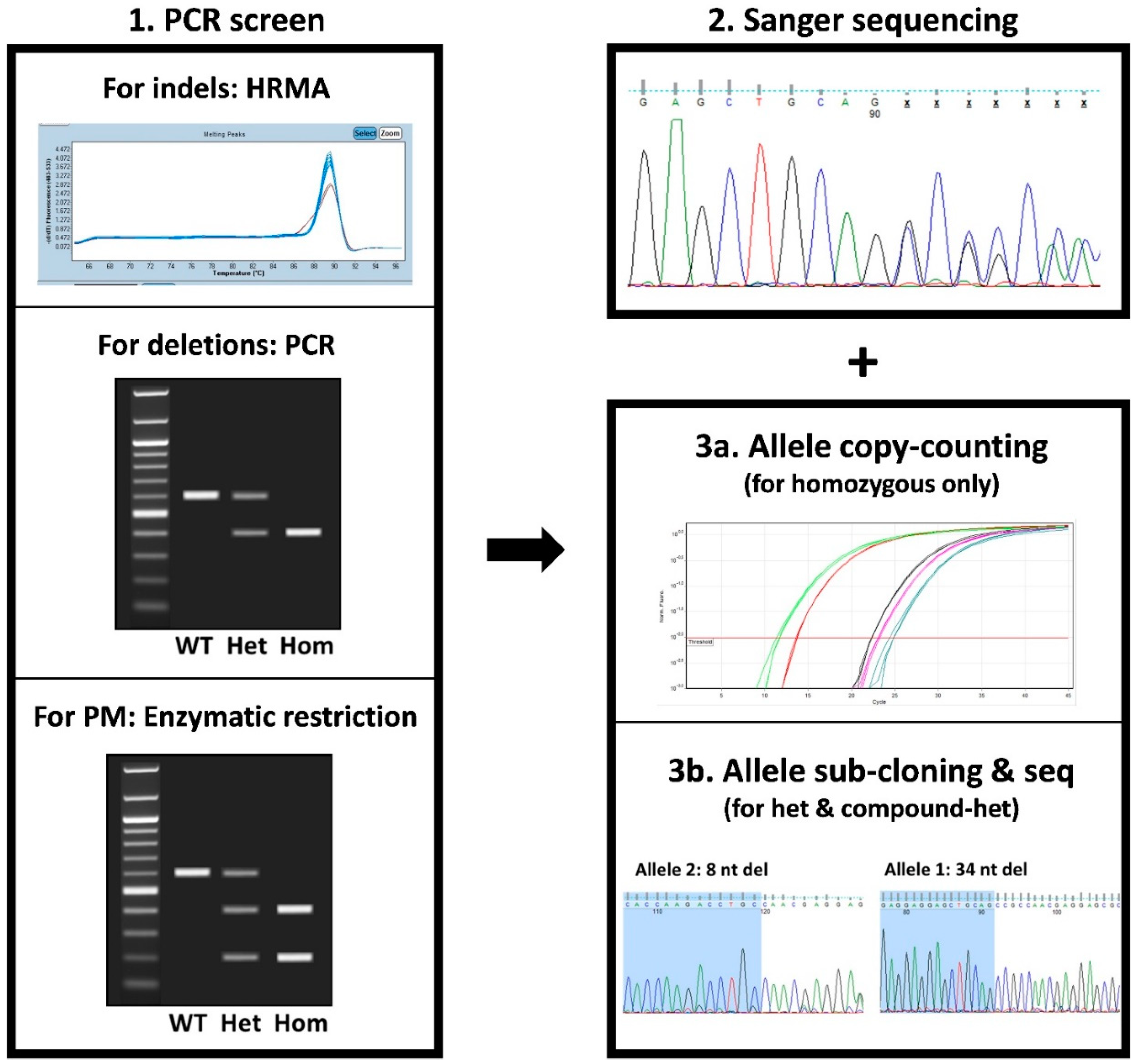
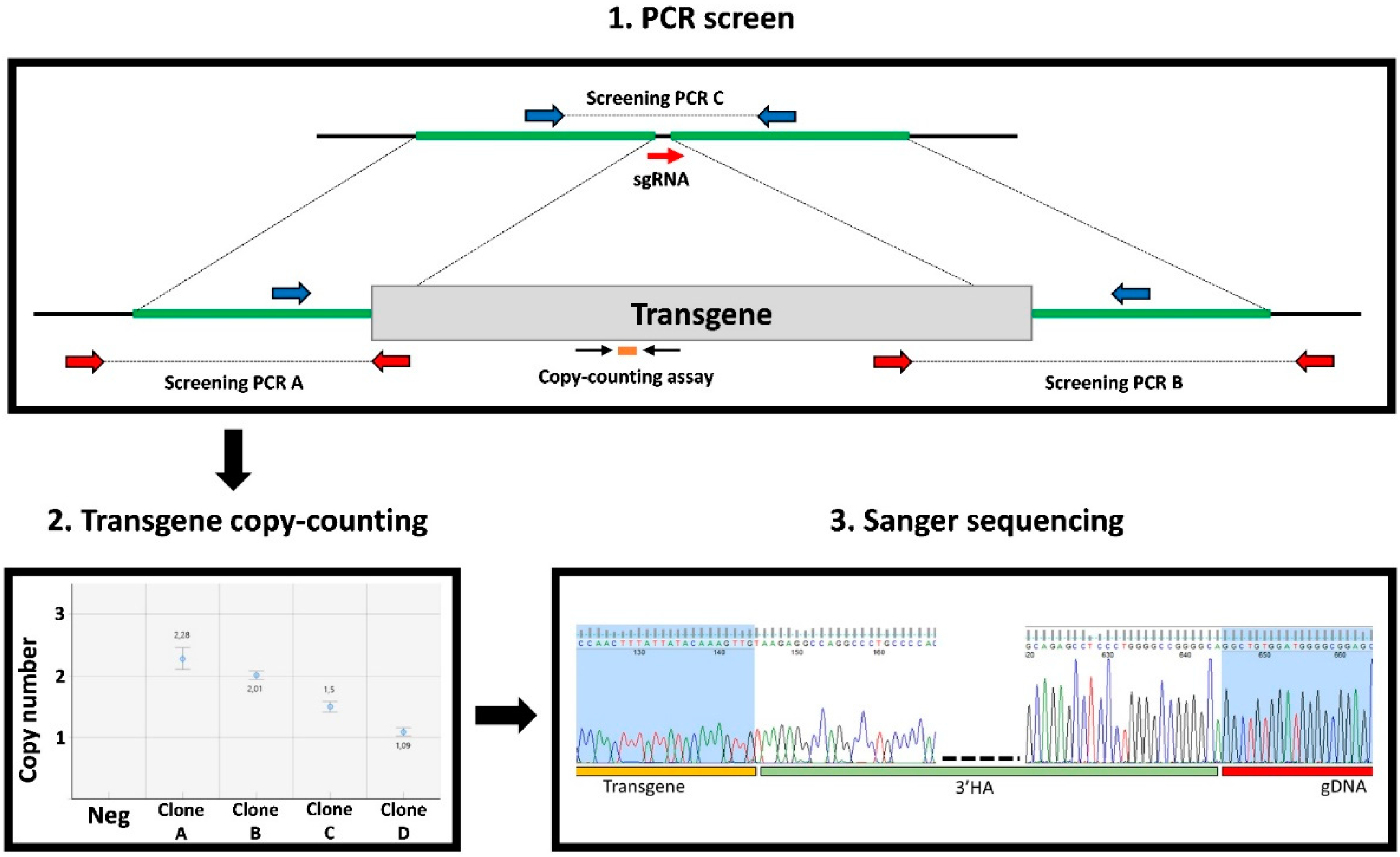
3.3. Clone Screening
3.4. In-Depth Molecular Characterization
4. Perspectives
Funding
Acknowledgments
Conflicts of Interest
References
- Thomson, J.A. Embryonic stem cell lines derived from human blastocysts. Science 1998, 282, 1145–1147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Mouse Embryonic and Adult Fibroblast Cultures by Defined Factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, Y.; Inoue, H.; Wu, J.C.; Yamanaka, S. Induced pluripotent stem cell technology: A decade of progress. Nat. Rev. Drug Discov. 2017, 16, 115–130. [Google Scholar] [CrossRef] [PubMed]
- Kilpinen, H.; Goncalves, A.; Leha, A.; Afzal, V.; Alasoo, K.; Ashford, S.; Bala, S.; Bensaddek, D.; Casale, F.P.; Culley, O.J.; et al. Common genetic variation drives molecular heterogeneity in human iPSCs. Nature 2017, 546, 370–375. [Google Scholar] [CrossRef] [Green Version]
- Bock, C.; Kiskinis, E.; Verstappen, G.; Gu, H.; Boulting, G.; Smith, Z.D.; Ziller, M.; Croft, G.F.; Amoroso, M.W.; Oakley, D.H.; et al. Reference maps of human es and ips cell variation enable high-throughput characterization of pluripotent cell lines. Cell 2011, 144, 439–452. [Google Scholar] [CrossRef] [Green Version]
- Hockemeyer, D.; Jaenisch, R. Induced pluripotent stem cells meet genome editing. Cell Stem Cell 2016, 18, 573–586. [Google Scholar] [CrossRef] [Green Version]
- Soldner, F.; Laganière, J.; Cheng, A.W.; Hockemeyer, D.; Gao, Q.; Alagappan, R.; Khurana, V.; Golbe, L.I.; Myers, R.H.; Lindquist, S.; et al. Generation of isogenic pluripotent stem cells differing exclusively at two early onset parkinson point mutations. Cell 2011, 146, 318–331. [Google Scholar] [CrossRef] [Green Version]
- Jang, Y.Y.; Ye, Z. Gene correction in patient-specific iPSCs for therapy development and disease modeling. Hum. Genet. 2016, 135, 1041–1058. [Google Scholar] [CrossRef] [Green Version]
- Ben Jehuda, R.; Shemer, Y.; Binah, O. Genome Editing in Induced Pluripotent Stem Cells using CRISPR/Cas9. Stem Cell Rev. Rep. 2018, 14, 323–336. [Google Scholar] [CrossRef]
- Li, X.-L.; Li, G.-H.; Fu, J.; Fu, Y.-W.; Zhang, L.; Chen, W.; Arakaki, C.; Zhang, J.-P.; Wen, W.; Zhao, M.; et al. Highly efficient genome editing via CRISPR–Cas9 in human pluripotent stem cells is achieved by transient BCL-XL overexpression. Nucleic Acids Res. 2018, 46, 1–21. [Google Scholar] [CrossRef]
- Mitzelfelt, K.A.; McDermott-Roe, C.; Grzybowski, M.N.; Marquez, M.; Kuo, C.T.; Riedel, M.; Lai, S.; Choi, M.J.; Kolander, K.D.; Helbling, D.; et al. Efficient Precision Genome Editing in iPSCs via Genetic Co-targeting with Selection. Stem Cell Rep. 2017, 8, 491–499. [Google Scholar] [CrossRef] [PubMed]
- Roberts, B.; Hendershott, M.C.; Arakaki, J.; Gerbin, K.A.; Malik, H.; Nelson, A.; Gehring, J.; Hookway, C.; Ludmann, S.A.; Yang, R.; et al. Fluorescent Gene Tagging of Transcriptionally Silent Genes in hiPSCs. Stem Cell Rep. 2019, 12, 1145–1158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberts, B.; Haupt, A.; Tucker, A.; Grancharova, T.; Arakaki, J.; Fuqua, M.A.; Nelson, A.; Hookway, C.; Ludmann, S.A.; Mueller, I.A.; et al. Systematic gene tagging using CRISPR/Cas9 in human stem cells to illuminate cell organization. Mol. Biol. Cell 2017, 28, 2854–2874. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Guell, M.; Byrne, S.; Yang, J.L.; De Los Angeles, A.; Mali, P.; Aach, J.; Kim-Kiselak, C.; Briggs, A.W.; Rios, X.; et al. Optimization of scarless human stem cell genome editing. Nucleic Acids Res. 2013, 41, 9049–9061. [Google Scholar] [CrossRef]
- Mianné, J.; Codner, G.F.; Caulder, A.; Fell, R.; Hutchison, M.; King, R.; Stewart, M.E.; Wells, S.; Teboul, L. Analysing the outcome of CRISPR-aided genome editing in embryos: Screening, genotyping and quality control. Methods 2017, 121–122, 68–76. [Google Scholar] [CrossRef]
- Canaj, H.; Hussmann, J.A.; Li, H.; Beckman, K.A.; Goodrich, L.; Cho, N.H.; Li, Y.J.; Santos, D.A.; McGeever, A.; Stewart, E.M.; et al. Deep profiling reveals substantial heterogeneity of integration outcomes in CRISPR knock-in experiments. bioRxiv 2019, 841098. [Google Scholar] [CrossRef] [Green Version]
- Hsu, P.D.; Lander, E.S.; Zhang, F. Development and applications of CRISPR-Cas9 for genome engineering. Cell 2014, 157, 1262–1278. [Google Scholar] [CrossRef] [Green Version]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef]
- Mitsunobu, H.; Teramoto, J.; Nishida, K.; Kondo, A. Beyond Native Cas9: Manipulating Genomic Information and Function. Trends Biotechnol. 2017, 35, 983–996. [Google Scholar] [CrossRef]
- Yeh, C.D.; Richardson, C.D.; Corn, J.E. Advances in genome editing through control of DNA repair pathways. Nat. Cell Biol. 2019, 21, 1468–1478. [Google Scholar] [CrossRef]
- Zhang, M.; Niibe, K.; Kondo, T.; Kamano, Y.; Saeki, M.; Egusa, H. Gene Delivery and Expression Systems in Induced Pluripotent Stem Cells. In Interface Oral Health Science 2016; Sasaki, K., Suzuki, O., Takahashi, N., Eds.; Springer: Singapore, 2017; pp. 121–133. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.; Kim, D.; Cho, S.W.; Kim, J.; Kim, J.S. Highly efficient RNA-guided genome editing in human cells via delivery of purified Cas9 ribonucleoproteins. Genome Res. 2014, 24, 1012–1019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Renaud, J.B.; Boix, C.; Charpentier, M.; De Cian, A.; Cochennec, J.; Duvernois-Berthet, E.; Perrouault, L.; Tesson, L.; Edouard, J.; Thinard, R.; et al. Improved Genome Editing Efficiency and Flexibility Using Modified Oligonucleotides with TALEN and CRISPR-Cas9 Nucleases. Cell Rep. 2016, 14, 2263–2272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Codner, G.F.; Mianné, J.; Caulder, A.; Loeffler, J.; Fell, R.; King, R.; Allan, A.J.; Mackenzie, M.; Pike, F.J.; McCabe, C.V.; et al. Application of long single-stranded DNA donors in genome editing: Generation and validation of mouse mutants. BMC Biol. 2018, 16, 70. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Beckman, K.A.; Pessino, V.; Huang, B.; Weissman, J.S.; Leonetti, M.D. Design and specificity of long ssDNA donors for CRISPR-based knock-in. bioRxiv 2017, 178905. [Google Scholar] [CrossRef] [Green Version]
- Yu, X.; Liang, X.; Xie, H.; Kumar, S.; Ravinder, N.; Potter, J.; de Mollerat du Jeu, X.; Chesnut, J.D. Improved delivery of Cas9 protein/gRNA complexes using lipofectamine CRISPRMAX. Biotechnol. Lett. 2016, 38, 919–929. [Google Scholar] [CrossRef] [Green Version]
- Timin, A.S.; Muslimov, A.R.; Lepik, K.V.; Epifanovskaya, O.S.; Shakirova, A.I.; Mock, U.; Riecken, K.; Okilova, M.V.; Sergeev, V.S.; Afanasyev, B.V.; et al. Efficient gene editing via non-viral delivery of CRISPR–Cas9 system using polymeric and hybrid microcarriers. Nanomed. Nanotechnol. Biol. Med. 2018, 14, 97–108. [Google Scholar] [CrossRef]
- Martin, R.M.; Ikeda, K.; Cromer, M.K.; Uchida, N.; Nishimura, T.; Romano, R.; Tong, A.J.; Lemgart, V.T.; Camarena, J.; Pavel-Dinu, M.; et al. Highly Efficient and Marker-free Genome Editing of Human Pluripotent Stem Cells by CRISPR-Cas9 RNP and AAV6 Donor-Mediated Homologous Recombination. Cell Stem Cell 2019, 24, 821–828. [Google Scholar] [CrossRef] [PubMed]
- Lino, C.A.; Harper, J.C.; Carney, J.P.; Timlin, J.A. Delivering CRISPR: A review of the challenges and approaches. Drug Deliv. 2018, 25, 1234–1257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, H.; Kauffman, K.J.; Anderson, D.G. Delivery technologies for genome editing. Nat. Rev. Drug Discov. 2017, 16, 387–399. [Google Scholar] [CrossRef] [PubMed]
- Scott, D.A.; Zhang, F. Implications of human genetic variation in CRISPR-based therapeutic genome editing. Nat. Med. 2017, 23, 1095–1101. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wang, Y.; Huang, H.; Chen, B.; Chen, X.; Hu, J.; Chang, T.; Lin, R.-J.; Yee, J.-K. Precise Gene Modification Mediated by TALEN and Single-Stranded Oligodeoxynucleotides in Human Cells. PLoS ONE 2014, 9, e93575. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M.; Parry-Smith, D.; Iyer, V. Best practice for CRISPR design using current tools and resources. Methods 2019, 164–165, 3–17. [Google Scholar] [CrossRef] [PubMed]
- Allen, F.; Crepaldi, L.; Alsinet, C.; Strong, A.J.; Kleshchevnikov, V.; De Angeli, P.; Páleníková, P.; Khodak, A.; Kiselev, V.; Kosicki, M.; et al. Predicting the mutations generated by repair of Cas9-induced double-strand breaks. Nat. Biotechnol. 2019, 37, 64–82. [Google Scholar] [CrossRef] [PubMed]
- Vouillot, L.; Thélie, A.; Pollet, N. Comparison of T7E1 and surveyor mismatch cleavage assays to detect mutations triggered by engineered nucleases. G3 Genes Genomes Genet. 2015, 5, 407–415. [Google Scholar] [CrossRef] [Green Version]
- Brinkman, E.K.; Chen, T.; Amendola, M.; Van Steensel, B. Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic Acids Res. 2014, 42, 1–8. [Google Scholar] [CrossRef]
- Zhang, J.P.; Li, X.L.; Li, G.H.; Chen, W.; Arakaki, C.; Botimer, G.D.; Baylink, D.; Zhang, L.; Wen, W.; Fu, Y.W.; et al. Efficient precise knockin with a double cut HDR donor after CRISPR/Cas9-mediated double-stranded DNA cleavage. Genome Biol. 2017, 18, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, K.; Tsunekawa, Y.; Hernandez-Benitez, R.; Wu, J.; Zhu, J.; Kim, E.J.; Hatanaka, F.; Yamamoto, M.; Araoka, T.; Li, Z.; et al. In vivo genome editing via CRISPR/Cas9 mediated homology-independent targeted integration. Nature 2016, 540, 144–149. [Google Scholar] [CrossRef]
- Oka, M.; Kobayashi, N.; Matsumura, K.; Nishio, M.; Saeki, K. Exogenous Cytokine-Free Differentiation of Human Pluripotent Stem Cells into Classical Brown Adipocytes. Cells 2019, 8, 373. [Google Scholar] [CrossRef] [Green Version]
- Braam, S.R.; Denning, C.; Van Den Brink, S.; Kats, P.; Hochstenbach, R.; Passier, R.; Mummery, C.L. Improved genetic manipulation of human embryonic stem cells. Nat. Methods 2008, 5, 389–392. [Google Scholar] [CrossRef]
- Watanabe, K.; Ueno, M.; Kamiya, D.; Nishiyama, A.; Matsumura, M.; Wataya, T.; Takahashi, J.B.; Nishikawa, S.; Nishikawa, S.I.; Muguruma, K.; et al. A ROCK inhibitor permits survival of dissociated human embryonic stem cells. Nat. Biotechnol. 2007, 25, 681–686. [Google Scholar] [CrossRef]
- Ihry, R.J.; Worringer, K.A.; Salick, M.R.; Frias, E.; Ho, D.; Theriault, K.; Kommineni, S.; Chen, J.; Sondey, M.; Ye, C.; et al. P53 inhibits CRISPR-Cas9 engineering in human pluripotent stem cells. Nat. Med. 2018, 24, 939–946. [Google Scholar] [CrossRef]
- Thomas, H.R.; Percival, S.M.; Yoder, B.K.; Parant, J.M. High-throughput genome editing and phenotyping facilitated by high resolution melting curve analysis. PLoS ONE 2014, 9. [Google Scholar] [CrossRef]
- Yang, Z.; Steentoft, C.; Hauge, C.; Hansen, L.; Thomsen, A.L.; Niola, F.; Vester-Christensen, M.B.; Frödin, M.; Clausen, H.; Wandall, H.H.; et al. Fast and sensitive detection of indels induced by precise gene targeting. Nucleic Acids Res. 2015, 43. [Google Scholar] [CrossRef] [Green Version]
- Findlay, S.D.; Vincent, K.M.; Berman, J.R.; Postovit, L.M. A digital pcr-based method for efficient and highly specific screening of genome edited cells. PLoS ONE 2016, 11, 1–17. [Google Scholar] [CrossRef]
- Kosicki, M.; Tomberg, K.; Bradley, A. Repair of double-strand breaks induced by CRISPR–Cas9 leads to large deletions and complex rearrangements. Nat. Biotechnol. 2018. [Google Scholar] [CrossRef]
- Mccabe, C.V.; Codner, G.F.; Allan, A.J.; Christou, S.; Loeffler, J.; Mackenzie, M.; Malzer, E.; Mianné, J.; Pike, F.J.; Hutchison, M.; et al. Application of long-read sequencing for robust identification of correct alleles in genome edited animals. bioRxiv 2019, 1–27. [Google Scholar] [CrossRef]
- Norris, A.L.; Lee, S.S.; Greenlees, K.J.; Tadesse, D.A.; Miller, M.F.; Lombardi, H.A. Template plasmid integration in germline genome-edited cattle. Nat. Biotechnol. 2020, 38, 163–164. [Google Scholar] [CrossRef]
- Haeussler, M. CRISPR off-targets: A question of context. Cell Biol. Toxicol. 2020, 36, 5–9. [Google Scholar] [CrossRef] [Green Version]
- Vakulskas, C.A.; Behlke, M.A. Evaluation and reduction of crispr off-target cleavage events. Nucleic Acid Ther. 2019, 29, 167–174. [Google Scholar] [CrossRef]
- Zhang, J.; Hirst, A.J.; Duan, F.; Qiu, H.; Huang, R.; Ji, Y.; Bai, L.; Zhang, F.; Robinson, D.; Jones, M.; et al. Anti-apoptotic Mutations Desensitize Human Pluripotent Stem Cells to Mitotic Stress and Enable Aneuploid Cell Survival. Stem Cell Rep. 2019, 12, 557–571. [Google Scholar] [CrossRef] [Green Version]
- Assou, S.; Bouckenheimer, J.; De Vos, J. Concise Review: Assessing the Genome Integrity of Human Induced Pluripotent Stem Cells: What Quality Control Metrics? Stem Cells 2018, 36, 814–821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Assou, S.; Girault, N.; Plinet, M.; Bouckenheimer, J.; Sansac, C.; Combe, M.; Mianne, J.; Bourguignon, C.; Fieldes, M.; Ahmed, E.; et al. Recurrent Genetic Abnormalities in Human Pluripotent Stem Cells: Definition and Routine Detection in Culture Supernatant by Targeted Droplet Digital PCR. Stem Cell Rep. 2020, 14, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Brookhouser, N.; Tekel, S.J.; Standage-Beier, K.; Nguyen, T.; Schwarz, G.; Wang, X.; Brafman, D.A. BIG-TREE: Base-Edited Isogenic hPSC Line Generation Using a Transient Reporter for Editing Enrichment. Stem Cell Rep. 2020, 14, 184–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anzalone, A.V.; Randolph, P.B.; Davis, J.R.; Sousa, A.A.; Koblan, L.W.; Levy, J.M.; Chen, P.J.; Wilson, C.; Newby, G.A.; Raguram, A.; et al. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature 2019, 576, 149–157. [Google Scholar] [CrossRef]
- Haake, K.; Ackermann, M.; Lachmann, N. Concise Review: Towards the Clinical Translation of Induced Pluripotent Stem Cell-Derived Blood Cells—Ready for Take-Off. Stem Cells Transl. Med. 2019, 8, 332–339. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Test (Reference) | Principle | Advantages | Limitations |
|---|---|---|---|
| T7E1 or Surveyor nuclease assay [35] | Enzymatic digestion of PCR heteroduplexes | Quick (few hours) Cheap Medium throughput Basic laboratory equipment | Sensitivity Medium throughput (needs also WT PCR product to detect homozygous clones) No information on the allele composition |
| HRMA [43] | Melting curve variation | Quick (few hours) Cheap High throughput Sensitive Basic laboratory equipment | Short amplicons required, might miss larger deletion events No information on the allele composition |
| IDAA [44] | Indel detection by amplicon analysis | Quick (few hours) High throughput Sensitive Information on the allele composition | More expensive Specific laboratory equipment |
| ddPCR [45] | Indel detection by drop-out of a labeled probe | Quick (few hours) Medium throughput Sensitive | More expensive Specific laboratory equipment No information on the allele composition |
| Test | Project Type | Purpose | Methods |
|---|---|---|---|
| DNA integrity | All | Identification of unwanted genomic abnormalities/rearrangements | Screening: qPCR or ddPCR Final characterization: G-banding/microarray/NGS |
| OT mutation | All | Identification of potential mutagenesis events at off-target sites | HRMA or PCR of the top 5-10 OT sites/HRMA or PCR of OT with up to 3 MMs/whole exome/whole genome |
| Pluripotency | All | Confirming the pluripotency state of the transgenic iPSC lines | IF/cytometry/embryoid bodies/teratoma |
| Plasmid integration | All projects in which plasmids are used | Confirming the absence of plasmid backbone integration in the clone’s genome | PCR/qPCR/ddPCR |
| ssDNA donor integration | All projects in which ssDNA are used | Identification of unintended integration of the ssDNA donor oligo(s) | qPCR/ddPCR |
| p53 function | All | Confirming p53 function | Sequencing |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mianné, J.; Bourguignon, C.; Nguyen Van, C.; Fieldès, M.; Nasri, A.; Assou, S.; De Vos, J. Pipeline for the Generation and Characterization of Transgenic Human Pluripotent Stem Cells Using the CRISPR/Cas9 Technology. Cells 2020, 9, 1312. https://doi.org/10.3390/cells9051312
Mianné J, Bourguignon C, Nguyen Van C, Fieldès M, Nasri A, Assou S, De Vos J. Pipeline for the Generation and Characterization of Transgenic Human Pluripotent Stem Cells Using the CRISPR/Cas9 Technology. Cells. 2020; 9(5):1312. https://doi.org/10.3390/cells9051312
Chicago/Turabian StyleMianné, Joffrey, Chloé Bourguignon, Chloé Nguyen Van, Mathieu Fieldès, Amel Nasri, Said Assou, and John De Vos. 2020. "Pipeline for the Generation and Characterization of Transgenic Human Pluripotent Stem Cells Using the CRISPR/Cas9 Technology" Cells 9, no. 5: 1312. https://doi.org/10.3390/cells9051312