Hinge and Transmembrane Domains of Chimeric Antigen Receptor Regulate Receptor Expression and Signaling Threshold


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Lines
2.2. Mice
2.3. Construction of CAR Structural Variants
2.4. Production of CAR-T Cells
2.5. Flow Cytometry Analysis for Surface Expression and Antigen-Binding Capacity of CAR
2.6. Reverse Transcription and Quantitative Polymerase Chain Reaction
2.7. Western Blot Analysis
2.8. Cytotoxicity Assay
2.9. BrdU Proliferation Assay and Cytokine Enzyme-Linked Immunosorbent Assay (ELISA)
2.10. Statistical Analysis
3. Results
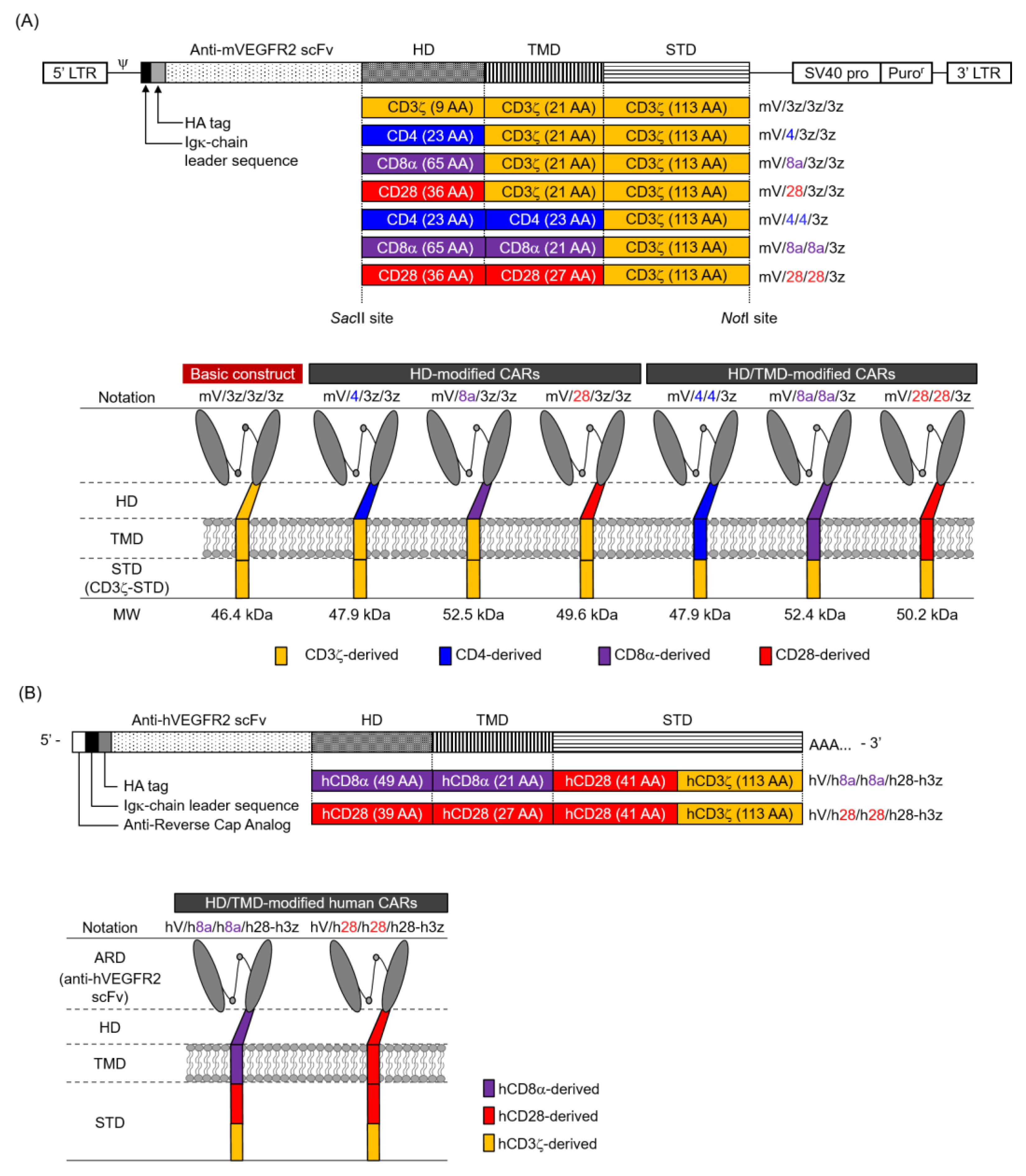
3.1. Expression of Various Hinge Domain (HD)-Modified and Hinge/Transmembrane Domain (HD/TMD)-Modified CARs in Mouse T Cells
3.2. Function of Mouse T Cells Expressing Various HD-Modified and HD/TMD-Modified CARs
3.3. Expression and Function of HD/TMD-Modified CARs in Human T Cells
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Restifo, N.P.; Dudley, M.E.; Rosenberg, S.A. Adoptive immunotherapy for cancer: Harnessing the T cell response. Nat. Rev. Immunol. 2012, 12, 269–281. [Google Scholar] [CrossRef]
- Tran, E.; Turcotte, S.; Gros, A.; Robbins, P.F.; Lu, Y.C.; Dudley, M.E.; Wunderlich, J.R.; Somerville, R.P.; Hogan, K.; Hinrichs, C.S.; et al. Cancer immunotherapy based on mutation-specific CD4+ T cells in a patient with epithelial cancer. Science 2014, 344, 641–645. [Google Scholar] [CrossRef]
- Eshhar, Z.; Waks, T.; Gross, G.; Schindler, D.G. Specific activation and targeting of cytotoxic lymphocytes through chimeric single chains consisting of antibody-binding domains and the gamma or zeta subunits of the immunoglobulin and T-cell receptors. Proc. Natl. Acad. Sci. USA 1993, 90, 720–724. [Google Scholar] [CrossRef] [Green Version]
- June, C.H.; Sadelain, M. Chimeric antigen receptor therapy. N. Engl. J. Med. 2018, 379, 64–73. [Google Scholar] [CrossRef]
- Li, J.; Li, W.; Huang, K.; Zhang, Y.; Kupfer, G.; Zhao, Q. Chimeric antigen receptor T cell (CAR-T) immunotherapy for solid tumors: Lessons learned and strategies for moving forward. J. Hematol. Oncol. 2018, 11, 22. [Google Scholar] [CrossRef] [Green Version]
- Maude, S.L.; Frey, N.; Shaw, P.A.; Aplenc, R.; Barrett, D.M.; Bunin, N.J.; Chew, A.; Gonzalez, V.E.; Zheng, Z.; Lacey, S.F.; et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N. Engl. J. Med. 2014, 371, 1507–1517. [Google Scholar] [CrossRef] [Green Version]
- Onea, A.S.; Jazirehi, A.R. CD19 chimeric antigen receptor (CD19 CAR)-redirected adoptive T-cell immunotherapy for the treatment of relapsed or refractory B-cell Non-Hodgkin’s Lymphomas. Am. J. Cancer Res. 2016, 15, 403–424. [Google Scholar]
- Maude, S.L.; Laetsch, T.W.; Buechner, J.; Rives, S.; Boyer, M.; Bittencourt, H.; Bader, P.; Verneris, M.R.; Stefanski, H.E.; Myers, G.D.; et al. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. N. Engl. J. Med. 2018, 378, 439–448. [Google Scholar] [CrossRef]
- Neelapu, S.S.; Locke, F.L.; Bartlett, N.L.; Lekakis, L.J.; Miklos, D.B.; Jacobson, C.A.; Braunschweig, I.; Oluwole, O.O.; Siddiqi, T.; Lin, Y.; et al. Axicabtagene Ciloleucel CAR T-cell therapy in refractory large B-cell lymphoma. N. Engl. J. Med. 2017, 377, 2531–2544. [Google Scholar] [CrossRef]
- Morgan, R.A.; Yang, J.C.; Kitano, M.; Dudley, M.E.; Laurencot, C.M.; Rosenberg, S.A. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol. Ther. 2010, 18, 843–851. [Google Scholar] [CrossRef]
- Kanagawa, N.; Yanagawa, T.; Mukai, Y.; Yoshioka, Y.; Okada, N.; Nakagawa, S. Tumor-targeting CTL expressing a single-chain Fv specific for VEGFR2. Biochem. Biophys. Res. Commun. 2010, 394, 54–58. [Google Scholar] [CrossRef] [PubMed]
- Kanagawa, N.; Yanagawa, T.; Nakagawa, T.; Okada, N.; Nakagawa, S. Tumor vessel-injuring ability improves antitumor effect of cytotoxic T lymphocytes in adoptive immunotherapy. Cancer Gene Ther. 2013, 20, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Inoo, K.; Inagaki, R.; Fujiwara, K.; Sasawatari, S.; Kamigaki, T.; Nakagawa, S.; Okada, N. Immunological quality and performance of tumor vessel-targeting CAR-T cells prepared by mRNA-EP for clinical research. Mol. Ther. Oncolytics. 2016, 3, 16024. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, K.; Masutani, M.; Tachibana, M.; Okada, N. Impact of scFv structure in chimeric antigen receptor on receptor expression efficiency and antigen recognition properties. Biochem. Biophys. Res. Commun. 2020, in press. [Google Scholar] [CrossRef]
- Kusabuka, H.; Fujiwara, K.; Tokunaga, Y.; Hirobe, S.; Nakagawa, S.; Okada, N. Highly efficient gene transfer using a retroviral vector into murine T cells for preclinical chimeric antigen receptor-expressing T cell therapy. Biochem. Biophys. Res. Commun. 2016, 473, 73–79. [Google Scholar] [CrossRef]
- Ono, S.; Ohno, H.; Saito, T. Rapid turnover of the CD3 zeta chain independent of the TCR-CD3 complex in normal T cells. Immunity 1995, 2, 639–644. [Google Scholar] [CrossRef] [Green Version]
- Moody, A.M.; Chui, D.; Reche, P.A.; Priatel, J.J.; Marth, J.D.; Reinherz, E.L. Developmentally regulated glycosylation of the CD8alphabeta coreceptor stalk modulates ligand binding. Cell 2001, 107, 501–512. [Google Scholar] [CrossRef] [Green Version]
- Lazar-Molnar, E.; Almo, S.C.; Nathenson, S.G. The interchain disulfide linkage is not a prerequisite but enhances CD28 costimulatory function. Cell Immunol. 2006, 244, 125–129. [Google Scholar] [CrossRef] [Green Version]
- Ma, B.Y.; Mikolajczak, S.A.; Yoshida, T.; Yoshida, R.; Kelvin, D.J.; Ochi, A. CD28 T cell costimulatory receptor function is negatively regulated by N-linked carbohydrates. Biochem. Biophys. Res. Commun. 2004, 317, 60–67. [Google Scholar] [CrossRef]
- Titov, A.; Valiullina, A.; Zmievskaya, E.; Zaikova, E.; Petukhov, A.; Miftakhova, R.; Bulatov, E.; Rizvanov, A. Advancing CAR T-Cell Therapy for Solid Tumors: Lessons Learned From Lymphoma Treatment. Cancers 2020, 12, 125. [Google Scholar] [CrossRef] [Green Version]
- Stoiber, S.; Cadilha, B.L.; Benmebarek, M.R.; Lesch, S.; Endres, S.; Kobold, S. Limitations in the design of chimeric antigen receptors for cancer therapy. Cells 2019, 8, 472. [Google Scholar] [CrossRef] [Green Version]
- Guedan, S.; Calderon, H.; Posey, A.D., Jr.; Maus, M.V. Engineering and design of chimeric antigen receptors. Mol. Ther. Methods Clin. Dev. 2018, 12, 145–156. [Google Scholar] [CrossRef] [Green Version]
- Luo, C.; Wei, J.; Han, W. Spotlight on chimeric antigen receptor engineered T cell research and clinical trials in China. Sci. China Life Sci. 2016, 59, 349–359. [Google Scholar] [CrossRef] [Green Version]
- Brudno, J.N.; Kochenderfer, J.N. Chimeric antigen receptor T-cell therapies for lymphoma. Nat. Rev. Clin. Oncol. 2018, 15, 31–46. [Google Scholar] [CrossRef]
- Hudecek, M.; Lupo-Stanghellini, M.T.; Kosasih, P.L.; Sommermeyer, D.; Jensen, M.C.; Rader, C.; Riddell, S.R. Receptor affinity and extracellular domain modifications affect tumor recognition by ROR1-specific chimeric antigen receptor T cells. Clin. Cancer Res. 2013, 19, 3153–3164. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Jiang, S.; Fang, C.; Yang, S.; Olalere, D.; Pequignot, E.C.; Cogdill, A.P.; Li, N.; Ramones, M.; Granda, B.; et al. Affinity-tuned ErbB2 or EGFR chimeric antigen receptor T cells exhibit an increased therapeutic index against tumors in mice. Cancer Res. 2015, 75, 3596–3607. [Google Scholar] [CrossRef] [Green Version]
- Drent, E.; Poels, R.; Ruiter, R.; van de Donk, N.; Zweegman, S.; Yuan, H.; de Bruijn, J.D.; Sadelain, M.; Lokhorst, H.M.; Groen, R.W.J.; et al. Combined CD28 and 4-1BB costimulation potentiates affinity-tuned chimeric antigen receptor–engineered T cells. Clin. Cancer Res. 2019, 25, 4014–4025. [Google Scholar] [CrossRef] [Green Version]
- Finney, H.M.; Akbar, A.N.; Lawson, A.D. Activation of resting human primary T cells with chimeric receptors: Costimulation from CD28, inducible costimulator, CD134, and CD137 in series with signals from the TCR zeta chain. J. Immunol. 2004, 172, 104–113. [Google Scholar] [CrossRef]
- Guedan, S.; Posey, A.D., Jr.; Shaw, C.; Wing, A.; Da, T.; Patel, P.R.; McGettigan, S.E.; Casado-Medrano, V.; Kawalekar, O.U.; Uribe-Herranz, M.; et al. Enhancing CAR T cell persistence through ICOS and 4-1BB costimulation. JCI Insight 2018, 3, 96976. [Google Scholar] [CrossRef] [Green Version]
- Ying, Z.; He, T.; Wang, X.; Zheng, W.; Lin, N.; Tu, M.; Xie, Y.; Ping, L.; Zhang, C.; Liu, W.; et al. Parallel comparison of 4-1BB or CD28 co-stimulated CD19-targeted CAR-T cells for B cell Non-Hodgkin’s lymphoma. Mol. Ther. Oncolytics 2019, 15, 60–68. [Google Scholar] [CrossRef] [Green Version]
- Lippincott-Schwartz, J.; Roberts, T.H.; Hirschberg, K. Secretory Protein Trafficking and Organelle Dynamics in Living Cells. Annu. Rev. Cell Dev. Biol. 2000, 16, 557–589. [Google Scholar] [CrossRef] [PubMed]
- Krug, C.; Birkholz, K.; Paulus, A.; Schwenkert, M.; Schmidt, P.; Hoffmann, N.; Hombach, A.; Fey, G.; Abken, H.; Schuler, G.; et al. Stability and activity of MCSP-specific chimeric antigen receptors (CARs) depend on the scFv antigen-binding domain and the protein backbone. Cancer Immunol. Immunother. 2015, 64, 1623–1635. [Google Scholar] [CrossRef] [PubMed]
- Finetti, F.; Paccani, S.R.; Riparbelli, M.G.; Giacomello, E.; Perinetti, G.; Pazour, G.J.; Rosenbaum, J.L.; Baldari, C.T. Intraflagellar transport is required for polarized recycling of the TCR/CD3 complex to the immune synapse. Nat. Cell. Biol. 2009, 11, 1332–1339. [Google Scholar] [CrossRef] [Green Version]
- Xu, C.; Gagnon, E.; Call, M.E.; Schnell, J.R.; Schwieters, C.D.; Carman, C.V.; Chou, J.J.; Wucherpfennig, K.W. Regulation of T cell receptor activation by dynamic membrane binding of the CD3epsilon cytoplasmic tyrosine-Based motif. Cell 2008, 135, 702–713. [Google Scholar] [CrossRef] [Green Version]
- Künkele, A.; Johnson, A.J.; Rolczynski, L.S.; Chang, C.A.; Hoglund, V.; Kelly-Spratt, K.S.; Jensen, M.C. Functional tuning of CARs reveals signaling threshold above which CD8+ CTL antitumor potency is attenuated due to cell Fas-FasL-dependent AICD. Cancer Immunol. Res. 2015, 3, 368–379. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, N.; Bajgain, P.; Sukumaran, S.; Ansari, S.; Heslop, H.E.; Rooney, C.M.; Brenner, M.K.; Leen, A.M.; Vera, J.F. Fine-tuning the CAR spacer improves T-cell potency. Oncoimmunology 2016, 5, e1253656. [Google Scholar] [CrossRef] [Green Version]
- Bridgeman, J.S.; Hawkins, R.E.; Bagley, S.; Blaylock, M.; Holland, M.; Gilham, D.E. The optimal antigen response of chimeric antigen receptors harboring the CD3zeta transmembrane domain is dependent upon incorporation of the receptor into the endogenous TCR/CD3 complex. J. Immunol. 2010, 184, 6938–6949. [Google Scholar] [CrossRef] [Green Version]
- Arch, R.H.; Thompson, C.B. 4-1BB and Ox40 Are Members of a Tumor Necrosis Factor (TNF)-nerve Growth Factor Receptor Subfamily That Bind TNF Receptor-Associated Factors and Activate Nuclear Factor kappaB. Mol. Cell Biol. 1998, 18, 558–565. [Google Scholar] [CrossRef] [Green Version]
- Eyquem, J.; Mansilla-Soto, J.; Giavridis, T.; van der Stegen, S.J.; Hamieh, M.; Cunanan, K.M.; Odak, A.; Gönen, M.; Sadelain, M. Targeting a CAR to the TRAC locus with CRISPR/Cas9 enhances tumour rejection. Nature 2017, 543, 113–117. [Google Scholar] [CrossRef] [Green Version]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fujiwara, K.; Tsunei, A.; Kusabuka, H.; Ogaki, E.; Tachibana, M.; Okada, N. Hinge and Transmembrane Domains of Chimeric Antigen Receptor Regulate Receptor Expression and Signaling Threshold. Cells 2020, 9, 1182. https://doi.org/10.3390/cells9051182
Fujiwara K, Tsunei A, Kusabuka H, Ogaki E, Tachibana M, Okada N. Hinge and Transmembrane Domains of Chimeric Antigen Receptor Regulate Receptor Expression and Signaling Threshold. Cells. 2020; 9(5):1182. https://doi.org/10.3390/cells9051182
Chicago/Turabian StyleFujiwara, Kento, Ayaka Tsunei, Hotaka Kusabuka, Erika Ogaki, Masashi Tachibana, and Naoki Okada. 2020. "Hinge and Transmembrane Domains of Chimeric Antigen Receptor Regulate Receptor Expression and Signaling Threshold" Cells 9, no. 5: 1182. https://doi.org/10.3390/cells9051182