ONX 0914 Lacks Selectivity for the Cardiac Immunoproteasome in CoxsackievirusB3 Myocarditis of NMRI Mice and Promotes Virus-Mediated Tissue Damage

 , and
, and
Abstract
:1. Introduction
2. Results
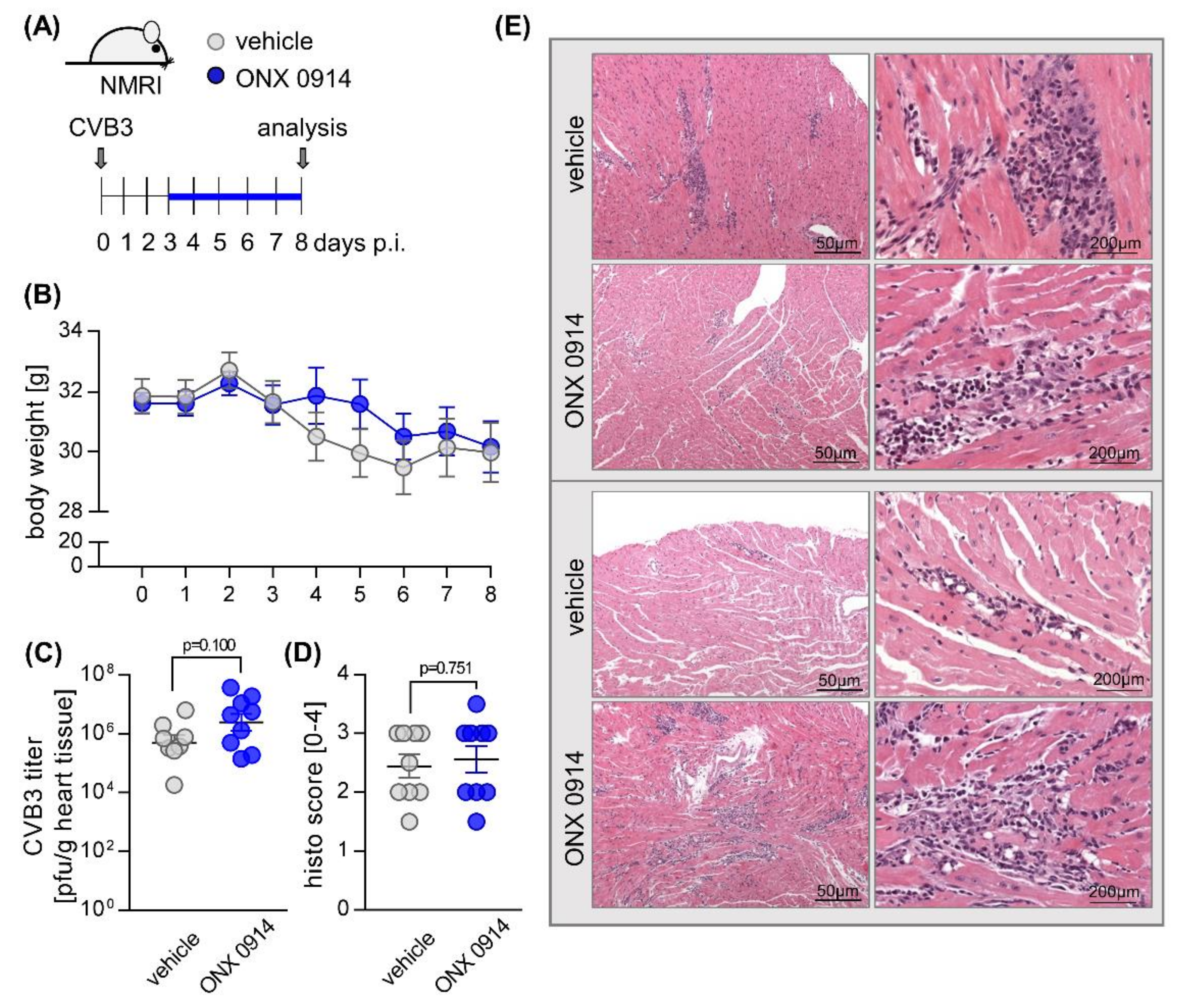
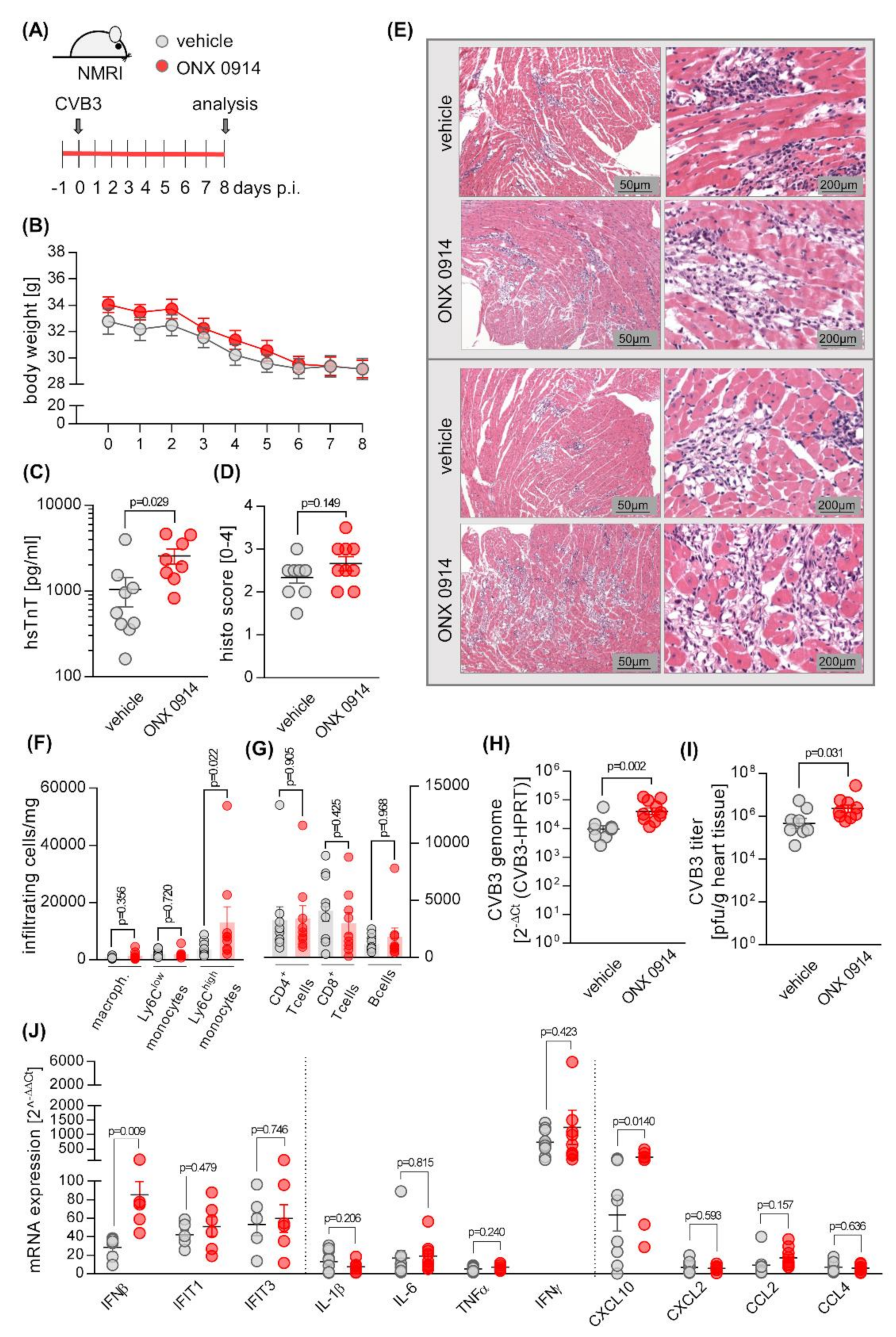
2.1. Influence of ONX 0914 on Myocarditis in NMRI Mice
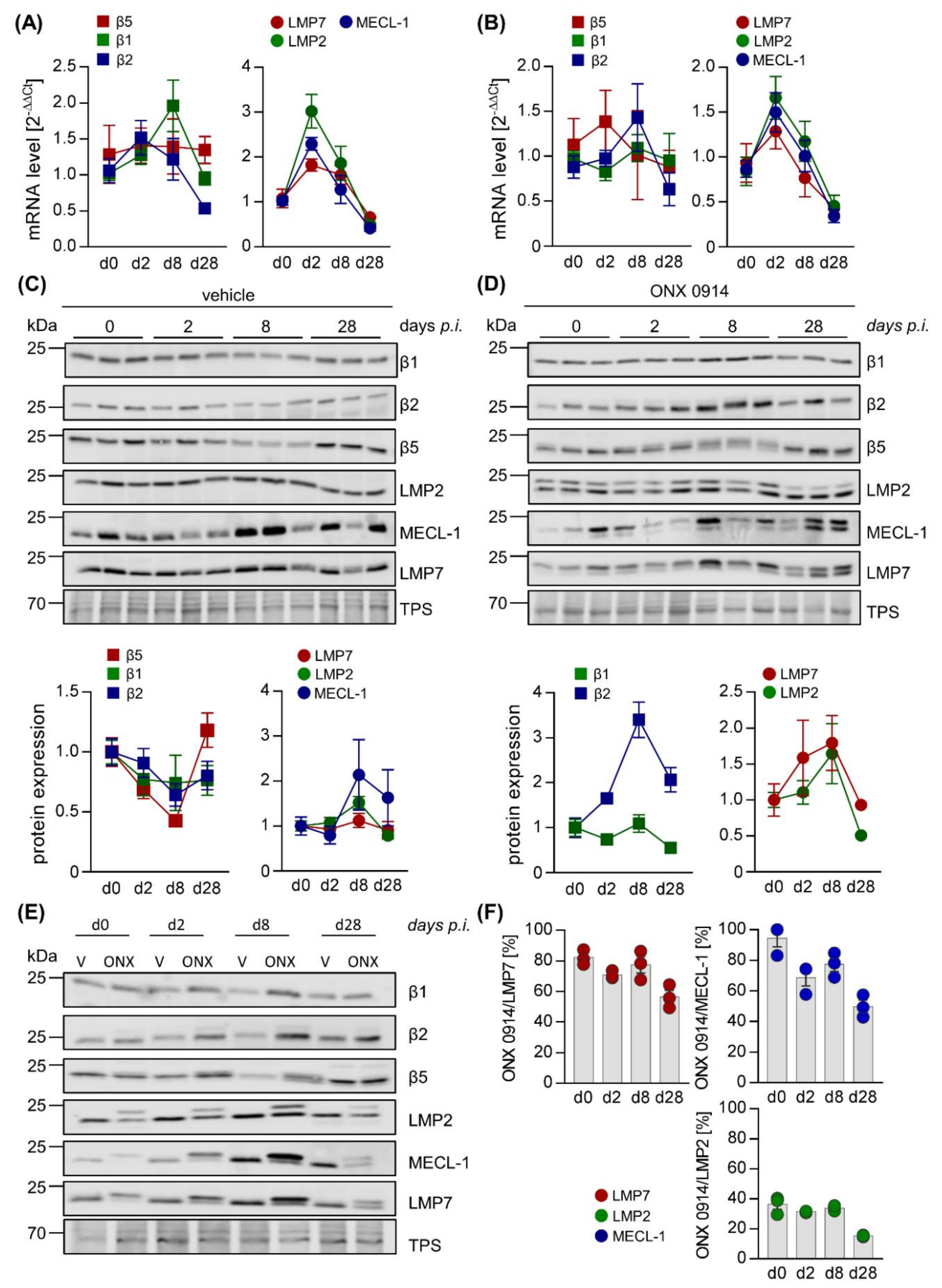
2.2. Influence of ONX 0914 on the Molecular Architecture of the 20S Proteasome Complex in Viral Myocarditis
3. Discussion
4. Materials and Methods
4.1. Mouse Studies
4.2. Echocardiography
4.3. Proteasome Inhibitor ONX 0914
4.4. Quantification of Infectious Viral Particles
4.5. Histology
4.6. Flow Cytometry
4.7. RNA Isolation, cDNA Synthesis, and qPCR
4.8. Western Blot Analysis
4.9. Statistics
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| bpm | beats per minute |
| CCL2 | chemokine ligand 2 |
| CCL4 | chemokine ligand 4 |
| CVB3 | coxsackievirus B3 |
| CXCL2 | C-X-C- motif chemokine ligand 2 |
| CXCL10 | C-X-C- motif chemokine ligand 10 |
| DAMP | damage-associated molecular pattern |
| EF | ejection fraction |
| HE | hematoxylin-eosin |
| IL | interleukin |
| IFIT1/3 | interferon-induced protein with tetratricopeptide repeats 1/3 |
| IFN | interferon |
| i-proteasome | immunoproteasome |
| LMP2 | low molecular weight protein 2 |
| LMP7 | low molecular weight protein 7 |
| LVID-d/-s | left ventricular inner dimension at diastole/systole |
| LCMV | lymphocytic choriomeningitis virus |
| MHC | major histocompatibility complex |
| mHPRT | mouse hypoxanthine-guanine phosphoribosyl transferase |
| MECL-1 | multicatalytic endopeptidase complex-like 1 |
| PAMP | pathogen-associated molecular pattern |
| PW-Doppler | pulsed-wave Doppler |
| (hs)TnT | (high sensitive) troponin T |
| UPS | ubiquitin-proteasome system |
| vol d/s | end-diastolic/end-systolic volume |
References
- Dikic, I. Proteasomal and Autophagic Degradation Systems. Annu. Rev. Biochem. 2017, 86, 193–224. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, A.L. Protein degradation and protection against misfolded or damaged proteins. Nature 2003, 426, 895–899. [Google Scholar] [CrossRef] [PubMed]
- Opitz, E.; Koch, A.; Klingel, K.; Schmidt, F.; Prokop, S.; Rahnefeld, A.; Sauter, M.; Heppner, F.L.; Völker, U.; Kandolf, R.; et al. Impairment of immunoproteasome function by beta5i/LMP7 subunit deficiency results in severe enterovirus myocarditis. PLoS Pathog. 2011, 7, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaczynska, M.; Rock, K.L.; Goldberg, A.L. Gamma-Interferon and Expression of Mhc Genes Regulate Peptide Hydrolysis by Proteasomes. Nature 1993, 365, 264–267. [Google Scholar] [CrossRef] [PubMed]
- Spur, E.-M.; Althof, N.; Respondek, D.; Klingel, K.; Heuser, A.; Overkleeft, H.S.; Voigt, A.; Beling, A. Inhibition of chymotryptic-like standard proteasome activity exacerbates doxorubicin-induced cytotoxicity in primary cardiomyocytes. Toxicology 2016, 353, 34–47. [Google Scholar] [CrossRef] [Green Version]
- Pickering, A.M.; Koop, A.L.; Teoh, C.Y.; Ermak, G.; Grune, T.; Davies, K.J. The immunoproteasome, the 20S proteasome and the PA28 alpha beta proteasome regulator are oxidative-stress-adaptive proteolytic complexes. Biochem. J. 2010, 432, 585–594. [Google Scholar] [CrossRef] [Green Version]
- Aki, M.; Shimbara, N.; Takashina, M.; Akiyama, K.; Kagawa, S.; Tamura, T.; Tanahashi, N.; Yoshimura, T.; Tanaka, K.; Ichihara, A. Interferon-γ Induces Different Subunit Organizations and Functional Diversity of Proteasomes. J. Biochem. 1994, 115, 257–269. [Google Scholar] [CrossRef]
- Jäkel, S.; Kuckelkorn, U.; Szalay, G.; Plötz, M.; Textoris-Taube, K.; Opitz, E.; Klingel, K.; Stevanovic, S.; Kandolf, R.; Kotsch, K.; et al. Differential Interferon Responses Enhance Viral Epitope Generation by Myocardial Immunoproteasomes in Murine Enterovirus Myocarditis. Am. J. Pathol. 2009, 175, 510–518. [Google Scholar] [CrossRef] [Green Version]
- Mishto, M.; Liepe, J.; Textoris-Taube, K.; Keller, C.; Henklein, P.; Weberruß, M.; Dahlmann, B.; Enenkel, C.; Voigt, A.; Kuckelkorn, U.; et al. Proteasome isoforms exhibit only quantitative differences in cleavage and epitope generation. Eur. J. Immunol. 2014, 44, 3508–3521. [Google Scholar] [CrossRef]
- Ebstein, F.; Voigt, A.; Lange, N.; Warnatsch, A.; Schröter, F.; Prozorovski, T.; Kuckelkorn, U.; Aktas, O.; Seifert, U.; Kloetzel, P.-M.; et al. Immunoproteasomes are important for proteostasis in immune responses. Cell 2013, 152, 935–937. [Google Scholar] [CrossRef] [Green Version]
- Seifert, U.; Bialy, L.; Ebstein, F.; Bech-Otschir, D.; Voigt, A.; Schröter, F.; Prozorovski, T.; Lange, N.; Steffen, J.; Rieger, M.; et al. Immunoproteasomes Preserve Protein Homeostasis upon Interferon-Induced Oxidative Stress. Cell 2010, 142, 613–624. [Google Scholar] [CrossRef] [Green Version]
- Huber, E.M.; Basler, M.; Schwab, R.; Heinemeyer, W.; Kirk, C.J.; Groettrup, M.; Groll, M. Immuno- and Constitutive Proteasome Crystal Structures Reveal Differences in Substrate and Inhibitor Specificity. Cell 2012, 148, 727–738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santos, R.D.L.A.; Bai, L.; Singh, P.K.; Murakami, N.; Fan, H.; Zhan, W.; Zhu, Y.; Jiang, X.; Zhang, K.; Assker, J.P.; et al. Structure of human immunoproteasome with a reversible and noncompetitive inhibitor that selectively inhibits activated lymphocytes. Nat. Commun. 2017, 8, 1692. [Google Scholar] [CrossRef] [PubMed]
- Gaczynska, M.; Rock, K.L.; Spies, T.; Goldberg, A.L. Peptidase activities of proteasomes are differentially regulated by the major histocompatibility complex-encoded genes for LMP2 and LMP. Proc. Natl. Acad. Sci. USA 1994, 91, 9213–9217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kincaid, E.Z.; Che, J.W.; York, I.; Escobar, H.; Reyes-Vargas, E.; Delgado, J.C.; Welsh, R.M.; Karow, M.L.; Murphy, A.J.; Valenzuela, D.M.; et al. Mice completely lacking immunoproteasomes show major changes in antigen presentation. Nat. Immunol. 2011, 13, 129–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tu, L.; Moriya, C.; Imai, T.; Ishida, H.; Tetsutani, K.; Duan, X.; Murata, S.; Tanaka, K.; Shimokawa, C.; Hisaeda, H.; et al. Critical role for the immunoproteasome subunit LMP7 in the resistance of mice to Toxoplasma gondii infection. Eur. J. Immunol. 2009, 39, 3385–3394. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Norbury, C.C.; Cho, Y.; Yewdell, J.W.; Bennink, J.R. Immunoproteasomes Shape Immunodominance Hierarchies of Antiviral Cd8+ T Cells at the Levels of T Cell Repertoire and Presentation of Viral Antigens. J. Exp. Med. 2001, 193, 1319–1326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nussbaum, A.K.; Rodriguez-Carreno, M.P.; Benning, N.; Botten, J.; Whitton, J.L. Immunoproteasome-deficient mice mount largely normal CD8+ T cell responses to lymphocytic choriomeningitis virus infection and DNA vaccination. J. Immunol. 2005, 175, 1153–1160. [Google Scholar] [CrossRef] [Green Version]
- Fehling, H.; Swat, W.; LaPlace, C.; Kühn, R.; Rajewsky, K.; Müller, U.; Von Boehmer, H. MHC class I expression in mice lacking the proteasome subunit LMP. Science 1994, 265, 1234–1237. [Google Scholar] [CrossRef]
- Muchamuel, T.; Basler, M.; Aujay, M.A.; Suzuki, E.; Kalim, K.W.; Lauer, C.; Sylvain, C.; Ring, E.R.; Shields, J.; Jiang, J.; et al. A selective inhibitor of the immunoproteasome subunit LMP7 blocks cytokine production and attenuates progression of experimental arthritis. Nat. Med. 2009, 15, 781–787. [Google Scholar] [CrossRef] [Green Version]
- Basler, M.; Lindstrom, M.M.; LaStant, J.J.; Bradshaw, J.M.; Owens, T.D.; Schmidt, C.; Maurits, E.; Tsu, C.; Overkleeft, H.S.; Kirk, C.J.; et al. Co-inhibition of immunoproteasome subunits LMP2 and LMP7 is required to block autoimmunity. EMBO Rep. 2018, 19, e46512. [Google Scholar] [CrossRef] [PubMed]
- Karreci, E.S.; Fan, H.; Uehara, M.; Mihali, A.B.; Singh, P.K.; Kurdi, A.T.; Solhjou, Z.; Riella, L.V.; Ghobrial, I.; Laragione, T.; et al. Brief treatment with a highly selective immunoproteasome inhibitor promotes long-term cardiac allograft acceptance in mice. Proc. Natl. Acad. Sci. USA 2016, 113, E8425–E8432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basler, M.; Li, J.; Groettrup, M. On the role of the immunoproteasome in transplant rejection. Immunogenetics 2018, 71, 263–271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kisselev, A.F.; Groettrup, M. Subunit specific inhibitors of proteasomes and their potential for immunomodulation. Curr. Opin. Chem. Boil. 2014, 23, 16–22. [Google Scholar] [CrossRef] [Green Version]
- Beling, A.; Kespohl, M. Proteasomal Protein Degradation: Adaptation of Cellular Proteolysis with Impact on Virus—And Cytokine-Mediated Damage of Heart Tissue During Myocarditis. Front. Immunol. 2018, 9. [Google Scholar] [CrossRef]
- Koerner, J.; Brunner, T.; Groettrup, M. Inhibition and deficiency of the immunoproteasome subunit LMP7 suppress the development and progression of colorectal carcinoma in mice. Oncotarget 2017, 8, 50873–50888. [Google Scholar] [CrossRef] [Green Version]
- Vachharajani, N.; Joeris, T.; Luu, M.; Hartmann, S.; Pautz, S.; Jenike, E.; Pantazis, G.; Prinz, I.; Hofer, M.J.; Steinhoff, U.; et al. Prevention of colitis-associated cancer by selective targeting of immunoproteasome subunit LMP. Oncotarget 2017, 8, 50447–50459. [Google Scholar] [CrossRef]
- Bockstahler, M.; Fischer, A.; Goetzke, C.C.; Neumaier, H.L.; Sauter, M.; Kespohl, M.; Müller, A.-M.; Meckes, C.; Salbach, C.; Schenk, M.; et al. Heart-Specific Immune Responses in an Animal Model of AutoimmuneRelated Myocarditis Mitigated by an Immunoproteasome Inhibitor and Genetic Ablation. Circulation 2020. [Google Scholar] [CrossRef] [Green Version]
- Basler, M.; Mundt, S.; Muchamuel, T.; Moll, C.; Jiang, J.; Groettrup, M.; Kirk, C.J. Inhibition of the immunoproteasome ameliorates experimental autoimmune encephalomyelitis. EMBO Mol. Med. 2014, 6, 226–238. [Google Scholar] [CrossRef]
- Li, J.; Koerner, J.; Basler, M.; Brunner, T.; Kirk, C.J.; Groettrup, M. Immunoproteasome inhibition induces plasma cell apoptosis and preserves kidney allografts by activating the unfolded protein response and suppressing plasma cell survival factors. Kidney Int. 2019, 95, 611–623. [Google Scholar] [CrossRef]
- Mundt, S.; Basler, M.; Buerger, S.; Engler, H.; Groettrup, M. Inhibiting the immunoproteasome exacerbates the pathogenesis of systemic Candida albicans infection in mice. Sci. Rep. 2016, 6, 19434. [Google Scholar] [CrossRef] [PubMed]
- Ersching, J.; Vasconcelos, J.R.; Ferreira, C.P.; Caetano, B.C.; Machado, A.V.; Bruna–Romero, O.; Baron, M.A.; Ferreira, L.R.P.; Cunha-Neto, E.; Rock, K.L.; et al. The Combined Deficiency of Immunoproteasome Subunits Affects Both the Magnitude and Quality of Pathogen- and Genetic Vaccination-Induced CD8+ T Cell Responses to the Human Protozoan Parasite Trypanosoma cruzi. PLOS Pathog. 2016, 12, e1005593. [Google Scholar] [CrossRef] [PubMed]
- Corsten, M.F.; Schroen, B.; Heymans, S. Inflammation in viral myocarditis: Friend or foe? Trends Mol. Med. 2012, 18, 426–437. [Google Scholar] [CrossRef] [PubMed]
- Kallewaard, N.L.; Zhang, L.; Chen, J.-W.; Guttenberg, M.; Sánchez, M.D.; Bergelson, J.M. Tissue-Specific Deletion of the Coxsackievirus and Adenovirus Receptor Protects Mice from Virus-Induced Pancreatitis and Myocarditis. Cell Host Microbe 2009, 6, 91–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klingel, K.; Hohenadl, C.; Canu, A.; Albrecht, M.; Seemann, M.; Mall, G.; Kandolf, R. Ongoing enterovirus-induced myocarditis is associated with persistent heart muscle infection: Quantitative analysis of virus replication, tissue damage, and inflammation. Proc. Natl. Acad. Sci. USA 1992, 89, 314–318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szalay, G.; Meiners, S.; Voigt, A.; Lauber, J.; Spieth, C.; Speer, N.; Sauter, M.; Kuckelkorn, U.; Zell, A.; Klingel, K.; et al. Ongoing Coxsackievirus Myocarditis Is Associated with Increased Formation and Activity of Myocardial Immunoproteasomes. Am. J. Pathol. 2006, 168, 1542–1552. [Google Scholar] [CrossRef] [Green Version]
- Pinkert, S.; Dieringer, B.; Klopfleisch, R.; Savvatis, K.; Van Linthout, S.; Pryshliak, M.; Tschöpe, C.; Klingel, K.; Kurreck, J.; Beling, A.; et al. Early Treatment of Coxsackievirus B3-Infected Animals with Soluble Coxsackievirus-Adenovirus Receptor Inhibits Development of Chronic Coxsackievirus B3 Cardiomyopathy. Circ. Hearth Fail. 2019, 12, e005250. [Google Scholar] [CrossRef]
- Althof, N.; Goetzke, C.C.; Kespohl, M.; Voss, K.; Heuser, A.; Pinkert, S.; Kaya, Z.; Klingel, K.; Beling, A. The immunoproteasome-specific inhibitor ONX 0914 reverses susceptibility to acute viral myocarditis. EMBO Mol. Med. 2018, 10, 200–218. [Google Scholar] [CrossRef]
- McCarthy, M.K.; Malitz, D.H.; Molloy, C.T.; Procario, M.C.; Greiner, K.E.; Zhang, L.; Wang, P.; Day, S.M.; Powell, S.R.; Weinberg, J.B. Interferon-Dependent Immunoproteasome Activity During Mouse Adenovirus Type 1 Infection. Virology 2016, 498, 57–68. [Google Scholar] [CrossRef]
- Voigt, A.; Rahnefeld, A.; Kloetzel, P.M.P.; Krueger, E. Cytokine-induced oxidative stress in cardiac inflammation and heart failure—How the ubiquitin proteasome system targets this vicious cycle. Front. Physiol. 2013, 4, 42. [Google Scholar] [CrossRef] [Green Version]
- Paeschke, A.; Possehl, A.; Klingel, K.; Voss, M.; Voss, K.; Kespohl, M.; Sauter, M.; Overkleeft, H.S.; Althof, N.; Garlanda, C.; et al. The immunoproteasome controls the availability of the cardioprotective pattern recognition molecule Pentraxin. Eur. J. Immunol. 2015, 46, 619–633. [Google Scholar] [CrossRef] [PubMed]
- Epelman, S.; Liu, P.P.; Mann, D.L. Role of innate and adaptive immune mechanisms in cardiac injury and repair. Nat. Rev. Immunol. 2015, 15, 117–129. [Google Scholar] [CrossRef] [PubMed]
- Gangaplara, A.; Massilamany, C.; Brown, D.M.; Delhon, G.; Pattnaik, A.K.; Chapman, N.; Rose, N.; Steffen, D.; Reddy, J. Coxsackievirus B3 infection leads to the generation of cardiac myosin heavy chain-α-reactive CD4 T cells in A/J mice. Clin. Immunol. 2012, 144, 237–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chow, L.H.; Gauntt, C.J.; McManus, B.M. Differential effects of myocarditic variants of Coxsackievirus B3 in inbred mice. A pathologic characterization of heart tissue damage. Lab. Investig. 1991, 64, 55–64. [Google Scholar] [PubMed]
- Merkle, I.; Tonew, M.; Glück, B.; Schmidtke, M.; Egerer, R.; Stelzner, A. Coxsackievirus B3-induced chronic myocarditis in outbred NMRI mice. J. Hum. Virol. 2000, 2, 369–379. [Google Scholar]
- Pappritz, K.; Savvatis, K.; Miteva, K.; Kerim, B.; Dong, F.; Fechner, H.; Muller, I.; Brandt, C.; López, B.; González, A.; et al. Immunomodulation by adoptive regulatory T-cell transfer improves Coxsackievirus B3-induced myocarditis. FASEB J. 2018, 32, 6066–6078. [Google Scholar] [CrossRef]
- Mundt, S.; Engelhardt, B.; Kirk, C.J.; Groettrup, M.; Basler, M. Inhibition and deficiency of the immunoproteasome subunit LMP7 attenuates LCMV-induced meningitis. Eur. J. Immunol. 2015, 46, 104–113. [Google Scholar] [CrossRef] [Green Version]
- Basler, M.; Dajee, M.; Moll, C.; Groettrup, M.; Kirk, C.J. Prevention of Experimental Colitis by a Selective Inhibitor of the Immunoproteasome. J. Immunol. 2010, 185, 634–641. [Google Scholar] [CrossRef] [Green Version]
- Ichikawa, H.T.; Conley, T.; Muchamuel, T.; Jiang, J.; Lee, S.; Owen, T.; Barnard, J.; Nevarez, S.; Goldman, B.I.; Kirk, C.J.; et al. Beneficial effect of novel proteasome inhibitors in murine lupus via dual inhibition of type I interferon and autoantibody-secreting cells. Arthr. Rheum. 2012, 64, 493–503. [Google Scholar] [CrossRef]
- Schwarz, K.; Van den Broek, M.; Kostka, S.; Kraft, R.; Soza, A.; Schmidtke, G.; Kloetzel, P.M.; Groettrup, M. Overexpression of the proteasome subunits LMP2, LMP7, and MECL-1, but not PA28 alpha/beta, enhances the presentation of an immunodominant lymphocytic choriomeningitis virus T cell epitope. J. Immunol. 2000, 2000. 165, 768–778. [Google Scholar] [CrossRef] [Green Version]
- Strehl, B.; Joeris, T.; Rieger, M.; Visekruna, A.; Textoris-Taube, K.; Kaufmann, S.H.; Kloetzel, P.-M.; Kuckelkorn, U.; Steinhoff, U. Immunoproteasomes are essential for clearance of Listeria monocytogenes in nonlymphoid tissues but not for induction of bacteria-specific CD8+ T cells. J. Immunol. 2006, 177, 6238–6244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kemball, C.C.; Harkins, S.; Whitton, J.L. Enumeration and Functional Evaluation of Virus-Specific CD4+ and CD8+ T Cells in Lymphoid and Peripheral Sites of Coxsackievirus B3 Infection. J. Virol. 2008, 82, 4331–4342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kemball, C.C.; Harkins, S.; Whitmire, J.K.; Flynn, C.T.; Feuer, R.; Whitton, J.L. Coxsackievirus B3 Inhibits Antigen Presentation In Vivo, Exerting a Profound and Selective Effect on the MHC Class I Pathway. PLOS Pathog. 2009, 5, e1000618. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Basler, M.; Alvarez, G.; Brunner, T.; Kirk, C.J.; Groettrup, M. Immunoproteasome inhibition prevents chronic antibody-mediated allograft rejection in renal transplantation. Kidney Int. 2018, 93, 670–680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, H. Interplay between the virus and the ubiquitin–proteasome system: Molecular mechanism of viral pathogenesis. Curr. Opin. Virol. 2016, 17, 1–10. [Google Scholar] [CrossRef]
- Chen, S.-C.; Chang, L.-Y.; Wang, Y.-W.; Chen, Y.-C.; Weng, K.-F.; Shih, S.-R.; Shih, H.-M. Sumoylation-promoted Enterovirus 71 3C Degradation Correlates with a Reduction in Viral Replication and Cell Apoptosis. J. Boil. Chem. 2011, 286, 31373–31384. [Google Scholar] [CrossRef] [Green Version]
- Basler, M.; Claus, M.; Klawitter, M.; Goebel, H.; Groettrup, M. Immunoproteasome Inhibition Selectively Kills Human CD14+ Monocytes and as a Result Dampens IL-23 Secretion. J. Immunol. 2019, 203, 1776–1785. [Google Scholar] [CrossRef]
- Singh, P.K.; Fan, H.; Jiang, X.; Shi, L.; Nathan, C.F.; Lin, G. Immunoproteasome beta5i-Selective Dipeptidomimetic Inhibitors. Chem. Med. Chem. 2016, 11, 2127–2131. [Google Scholar] [CrossRef]
- Stewart, A.K.; Rajkumar, S.V.; Dimopoulos, M.A.; Masszi, T.; Špička, I.; Oriol, A.; Hájek, R.; Rosiñol, L.; Siegel, D.S.; Mihaylov, G.G.; et al. Carfilzomib, Lenalidomide, and Dexamethasone for Relapsed Multiple Myeloma. N. Engl. J. Med. 2015, 372, 142–152. [Google Scholar] [CrossRef]
- Richardson, P.G.; Sonneveld, P.; Schuster, M.W.; Irwin, D.; Stadtmauer, E.A.; Facon, T.; Harousseau, J.-L.; Ben-Yehuda, D.; Lonial, S.; Goldschmidt, H.; et al. Bortezomib or High-Dose Dexamethasone for Relapsed Multiple Myeloma. New Engl. J. Med. 2005, 352, 2487–2498. [Google Scholar] [CrossRef] [Green Version]
- Hasinoff, B.B.; Patel, D. Myocyte-Damaging Effects and Binding Kinetics of Boronic Acid and Epoxyketone Proteasomal-Targeted Drugs. Cardiovasc. Toxicol. 2018, 18, 557–568. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Collumn Name | Vehicle | ONX 0914 | ||
|---|---|---|---|---|
| Baseline | Day 28 | Baseline | Day 28 | |
| Heart rate (bpm) | 417 ± 18 | 440 ± 18 | 440 ± 18 | 426 ± 14 |
| Trace EF (%) | 46 ± 2.5 | 48.1 ± 2.9 | 43.6 ± 2.6 | 42.7 ± 2.0 |
| Cardiac output (mL/min) | 13.6 ± 1.0 | 14.6 ± 1.0 | 14.3 ± 1.1 | 14.0 ± 0.8 |
| Stroke volume (µL) | 32.4 ± 1.8 | 32.7 ± 1.5 | 32.0 ± 1.9 | 33.0 ± 1.8 |
| Vol d (µL) | 70.5 ± 1.7 | 70.0 ± 3.1 | 74.2 ± 3 | 77.0 ± 2.2 |
| Vol s (µL) | 38.1 ± 1.9 | 37.3 ± 3.0 | 42.2 ± 2.9 | 44.2 ± 2.0 |
| LVID-d (mm) | 4.3 ± 0.1 | 4.3 ± 0.1 | 4.3 ± 0.1 | 4.5 ± 0.1 |
| LVID-s (mm) | 3.2 ± 0.1 | 3.1 ± 0.1 | 3.2 ± 0.1 | 3.5 ± 0.1 |
| Vehicle | ONX 0914 | |||
|---|---|---|---|---|
| Baseline | Day 8 | Baseline | Day 8 | |
| Heart rate (bpm) | 427 ± 16 | 393 ± 16 | 500 ± 13 * | 405 ± 23 § |
| Trace EF (%) | 50.7 ± 1.7 | 48.4 ± 2.6 | 57.4 ± 2.3 | 49.1 ± 3.2 § |
| Cardiac output (mL/min) | 14.4 ± 1.2 | 10.0 ± 0.9 § | 18.6 ± 1.2 * | 12.1 ± 1.3 § |
| Stroke volume (µL) | 33.6 ± 2.1 | 25.2 ± 1.4 § | 37.3 ± 2.4 | 29.6 ± 2.3 § |
| Vol d (µL) | 66.5 ± 4.0 | 52.8 ± 3.0 § | 65.5 ± 4.0 | 60.3 ± 2.6 |
| Vol s (µL) | 33.0 ± 2.4 | 27.6 ± 2.6 | 28.1 ± 2.5 | 30.7 ± 2.3 |
| LVID-d (mm) | 4.2 ± 0.1 | 4.0 ± 0.1 | 4.2 ± 0.1 | 4.2 ± 0.1 |
| LVID-s (mm) | 3.1 ± 0.1 | 3.0 ± 0.1 | 2.9 ± 0.1 | 3.2 ± 0.1 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Neumaier, H.L.; Harel, S.; Klingel, K.; Kaya, Z.; Heuser, A.; Kespohl, M.; Beling, A. ONX 0914 Lacks Selectivity for the Cardiac Immunoproteasome in CoxsackievirusB3 Myocarditis of NMRI Mice and Promotes Virus-Mediated Tissue Damage. Cells 2020, 9, 1093. https://doi.org/10.3390/cells9051093
Neumaier HL, Harel S, Klingel K, Kaya Z, Heuser A, Kespohl M, Beling A. ONX 0914 Lacks Selectivity for the Cardiac Immunoproteasome in CoxsackievirusB3 Myocarditis of NMRI Mice and Promotes Virus-Mediated Tissue Damage. Cells. 2020; 9(5):1093. https://doi.org/10.3390/cells9051093
Chicago/Turabian StyleNeumaier, Hannah Louise, Shelly Harel, Karin Klingel, Ziya Kaya, Arnd Heuser, Meike Kespohl, and Antje Beling. 2020. "ONX 0914 Lacks Selectivity for the Cardiac Immunoproteasome in CoxsackievirusB3 Myocarditis of NMRI Mice and Promotes Virus-Mediated Tissue Damage" Cells 9, no. 5: 1093. https://doi.org/10.3390/cells9051093