1. Introduction
Proximity-dependent labeling methods for identifying protein–protein interactions (PPIs) have become an established class of tools developed and utilized by hundreds of researchers within the last decade. These methods include antibody-based approaches such as SPLAAT and EMARS [
1,
2], the ascorbate peroxidase-based approach called APEX [
3,
4], the biotin-ligase approach BioID [
5,
6], and, more recently, a pupylation-based approach termed PUP-IT for identifying membrane protein interactions [
7]. While these systems have many similarities, their respective limitations restrict their practical applications. For example, the accuracy of the SPLAAT and EMARS methods will heavily depend on the quality of the antibody used and is traditionally restricted to plasma membrane interactions. Recent modifications to these antibody-based approaches has expanded the use of primary antibodies in conjunction with HRP-conjugated secondary antibodies for proximity labeling within the nucleus of fixed cells [
8]. The APEX system utilizes an extremely rapid peroxidase-based labeling induced by exposure of cells or tissues to mM levels of H
2O
2 in cells preincubated with biotin-phenol and typically requires the use of SILAC to interpret the data due to high background labeling. BioID requires a long labeling period of 15–18 h and does not function well at temperatures below 37 °C, and PUP-IT may not be suitable for interactions within organelles since Pup, a prokaryotic 64-amino acid protein, cannot diffuse across membranes.
We developed and applied the original biotin ligase used for proximity-dependent labeling using a humanized version of the BirA protein from
E. coli with a R118G mutation [
6] that was known to enable promiscuous biotinylation [
9,
10]. Wild-type BirA selectively biotinylates acetyl coA carboxylase by releasing a primed bioAMP molecule for covalent attachment to a specific lysine [
11,
12]. The R118G mutation decreases the affinity of BirA* (hereafter referred to as BioID) for both biotin and bioAMP, about 40- and 440-fold compared to the wild-type, respectively [
11]. The reduced affinity to bioAMP leads to a dramatically enhanced release of reactive bioAMP molecules from the ligase which covalently labels available lysines on proteins which we demonstrated to occur within a ~10 nm radius [
2,
6,
9,
10,
13]. Therefore, BioID can be used to map protein networks within live cells. The reduced affinity of the BioID ligase to biotin likely prevents substantive biotinylation without the addition of excess levels of biotin (5–50 μM), thus enabling the ability to induce the onset of biotinylation and thus temporally control the promiscuous labeling to enable selective or comparative studies. We developed a second promiscuous biotin ligase (BioID2) from
A. aeolicus as a smaller, somewhat more biotin-sensitive alternative to the original BioID ligase [
5]. Overall, the BioID method has been cited and/or applied in over 300 articles investigating a wide range of proteins and subcellular domains (for review, see [
14,
15]).
While BioID/BioID2 have been applied successfully in many model systems including in live cultured cells, yeast [
16,
17], parasites [
18,
19,
20,
21,
22,
23,
24], plants [
25,
26], and mice [
27,
28,
29,
30], most experiments have been performed utilizing a 12–24 h labeling period, with few exceptions labeling for 1 h or 3 h [
31,
32]. Experiments requiring shorter labeling periods require a faster version of BioID and one that would work well at temperatures well below 37 °C. Two groups have reported versions of BioID that address some or all of these limitations. A
B. subtilis-based version of BioID [
33], called BASU, that was used to map RNA–protein interactions was originally claimed to be 1000 times faster than the original BioID, but these results have not been reproducible [
34]. More recently, a directed-evolution variant of BioID, called TurboID, was reported to be substantially faster than the original BioID ligase and capable of functioning effectively at lower temperatures [
34]. In that same study, a smaller version of TurboID (termed miniTurbo) was also developed by removing the N-terminus of TurboID. Here, we comparatively evaluate the practical application and labeling radius of TurboID and the original BioID. We use previously validated baits in the nuclear envelope—including Lamin A (LaA), Nup43, and Nup53—in order to reveal optimal practical applications and to better characterize the growing toolbox of proximity-dependent labeling biotin ligases [
6,
13].
2. Materials and Methods
2.1. Plasmids
3xHA-TurboID and 3xHA-miniTurbo were amplified via PCR from Addgene constructs #107171 and #107172, respectively. Amplified PCR products were inserted into pBabe via In-Fusion Recombination into mycBioID pBabe (Addgene #80901), replacing mycBioID, using EcoRI and XhoI restriction enzyme (RE) sites. LaA, Nup43, Nup53, and Sun2 were amplified by PCR from human cDNA. The PCR products were inserted into the newly made N-terminal 3xHA-TurboID using XhoI and PmeI or C-terminal TurboID-3xHA pBabe puro constructs using NaeI and EcoRI RE sites. 3xHA-BioID was made by amplifying 3xHA from TurboID-3xHA and BioID from mycBioID pBabe puro and two-step In-Fusion cloning. Amplified PCR product was inserted into pBabe puro using EcoRI and XhoI. C-terminal LaA or Nup53 were subsequently inserted using XhoI and PmeI restriction sites. BioID-3xHA was made by cutting out TurboID from –TurboID-3xHA pBabe with EcoRI and BamHI and inserting BioID amplified PCR product. N-terminal fusions of Nup43 or Sun2 were inserted via NaeI and EcoRI RE sites. Intermediate TurboID generations were synthesized at Gene Universal (Newark, DE, USA), PCR amplified, and inserted into 3xHA-BioID pBabe puro by removing BioID with AgeI and XhoI. Dox-inducible 3xHA-TurboID pRetroX pTight construct was made by amplifying 3xHA-TurboID and inserting it into pRetroX pTight via In-Fusion cloning into EcoRI and BamHI sites.
2.2. Cell Culture
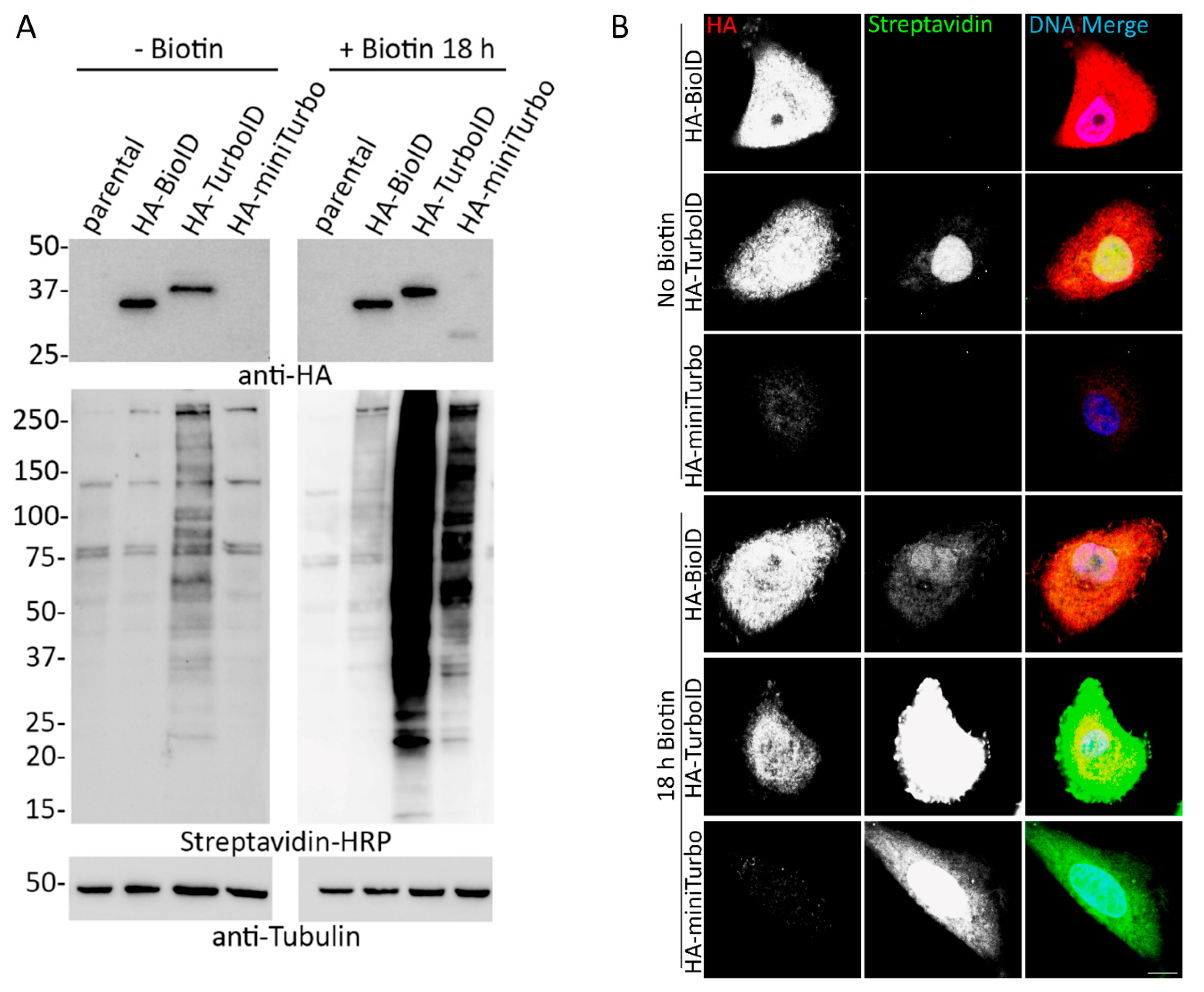
A549 cells were obtained from the American Type Culture Collection (ATCC; CCL-185™). Stable cell lines for all BioID, TurboID, and miniTurbo constructs were generated using retroviral transduction. HEK293 Phoenix cells (National Gene Vector Biorepository, Indianapolis, IN, USA) were transfected with each construct using Lipofectamine 3000 (Thermo Fisher Scientific, Waltham, MA, USA) per the manufacturer’s recommendation. The transfected cells were incubated at 37 °C for 6 h. After 6 h incubation, the transfected cells were replenished with fresh medium and further incubated at 32 °C for 72 h. The culture media was filtered through a 0.45-μm filter and added to A549 cells along with Polybrene (4 μg/mL; Santa Cruz Biotechnology, Dallas, TX, USA). At 72 h after transduction, puromycin (0.5 μg/mL; Thermo Fisher Scientific) was added to the target cells. The expression of fusion proteins and functional biotinylation was further verified using IF and WB. For cell lines requiring doxycycline (dox)-induction, dox (1 mg/mL) was added either 24 h before supplementation with 50 µM biotin (0 min, 10 min, 1 h, 4 h) or concurrently with 50 µmM biotin (18 h). Dialyzed serum was obtained from Fisher Scientific (Pittsburgh, PA, USA; Gibco, A3382001). The stable cell lines were maintained in 5.0% CO2 at 37 °C in DMEM (HyClone, Logan, UT, USA) supplemented with 10% fetal bovine serum (FBS). All cells were tested monthly for mycoplasma contamination.
2.3. Immunofluorescence
Cells grown on glass coverslips were fixed in 3% (wt/vol) paraformaldehyde/phosphate-buffered saline (PBS) for 10 min and permeabilized by 0.4% (wt/vol) Triton X-100/PBS for 15 min. For labeling fusion protein purposes, a mouse anti-hemagglutinin primary antibody was used (HA; 1:1000; 12CA5; Covance, Princeton, NJ, USA). The primary antibody was detected using Alexa Fluor 488–conjugated goat anti-mouse (1:1000; A11029; Thermo Fisher Scientific, Waltham, MA, USA) or Alexa Fluor 568–conjugated goat anti-mouse (1:1000; A11004; Thermo Fisher Scientific, Waltham, MA, USA). Alexa Fluor 488–conjugated streptavidin (S32354; Thermo Fisher Scientific, Waltham, MA, USA) or Alexa Fluor 568-conjugated streptavidin (S11226; Thermo Fisher Scientific, Waltham, MA, USA) was used to detect biotinylated proteins. DNA was detected with Hoechst dye 33342. Coverslips were mounted using 10% (wt/vol) Mowiol 4-88 (Polysciences, Inc., Warrington, PA, USA). Confocal images were obtained using a Nikon A1 confocal microscope (60 ×/1.49 oil APO TIRF Nikon objective) with a charge-coupled device camera (CoolSnap HQ; Photometrics, Tucson, AZ, USA) linked to a workstation running NIS-Elements software (Nikon, Melville, NY, USA). Epifluorescence images were captured using a Nikon Eclipse NiE (20 ×/0.75 Plan Apo Nikon objective) microscope.
2.4. Western Blot Analysis
To analyze total cell lysates by immunoblot, 1.2 × 106 cells were lysed in SDS–PAGE sample buffer, boiled for 5 min, and sonicated to shear DNA. Proteins were separated on 4–20% gradient gels (Mini-PROTEAN TGX; Bio-Rad, Hercules, CA, USA) and transferred to nitrocellulose membrane (Bio-Rad, Hercules, CA, US). After blocking with 10% (vol/vol) adult bovine serum and 0.2% Triton X-100 in PBS for 30 min, the membrane was incubated with appropriate primary antibodies: rabbit polyclonal anti-hemagglutinin (1:2000; Ab9110; Abcam, Cambridge, UK) and mouse monoclonal anti-tubulin as a loading control (1:10000; sc-32293; Santa Cruz Biotechnology, Dallas, TX, USA). The primary antibodies were detected using horseradish peroxidase (HRP)–conjugated anti-rabbit (1:40,000; G21234; Thermo Fisher Scientific, Waltham, MA, USA) or anti-mouse (1:40,000; F21453; Thermo Fisher Scientific, Waltham, MA, USA) antibodies. The signals from antibodies were detected using enhanced chemiluminescence via a Bio-Rad ChemiDoc MP System (Bio-Rad, Hercules, CA, USA). Following detection of each antibody, the membrane was quenched with 30% H2O2 for 30 min. To detect biotinylated proteins, the membrane was incubated with HRP-conjugated streptavidin (1:40,000; ab7403; Abcam, Cambridge, UK) in 0.2% Triton X-100 in PBS for 45 min.
2.5. BioID Pulldowns
Large-scale BioID pulldowns were performed as described in [
35]. For large-scale TurboID pulldowns, 2 10-cm dishes at 80% confluency were incubated with 50 μM biotin for 18 h and 2 10-cm dishes at 100% confluency were incubated with 50 μM biotin for either 0 or 10 min. From there, pulldowns were performed as described in [
35]. Briefly, cells were lysed in 8 M urea 50 mM Tris pH 7.4 containing protease inhibitor (87785: Thermo Fisher Scientific, Waltham, MA, USA) and DTT, incubated with universal nuclease (88700: Thermo Fisher Scientific, Waltham, MA, USA), and sonicated to further shear DNA. Lysates were precleared with Gelatin Sepharose 4B beads (17095601; GE Healthcare, Chicago, IL, USA) for 2 h and then incubated with Streptavidin Sepharose High Performance beads (17511301: GE Healthcare, Chicago, IL, USA) for 4 h. Streptavidin beads were washed four times with 8 M urea 50 mM Tris pH 7.4 wash buffer and resuspended in 50 mM ammonium bicarbonate with 1 mM biotin.
2.6. Protein Digestion
Beads were thawed and resuspended with 8 M urea, 50 mM ammonium bicarbonate, and cysteine disulfide bonds were reduced with 10 mM tris(2-carboxyethyl)phosphine (TCEP) at 30 °C for 60 min and cysteines were then alkylated with 30 mM iodoacetamide (IAA) in the dark at room temperature for 30 min. Following alkylation, urea was diluted to 1 M urea, and proteins were subjected to overnight digestion with mass spec grade Trypsin/Lys-C mix (Promega, Madison, WI, USA). Finally, beads were pulled down and the solution with peptides collected into a new tube. The beads were then washed once with 50 mM ammonium bicarbonate to increase peptide recovery.
Following digestion, samples were acidified with formic acid (FA) and subsequently desalted using AssayMap C18 cartridges (Agilent, Santa Clara, CA, USA) mounted on an Agilent AssayMap BRAVO liquid handling system. Briefly, C18 cartridges were first conditioned with 100% acetonitrile (ACN), followed 0.1% FA. Sample was then loaded onto the conditioned C18 cartridge, washed with 0.1% FA, and eluted with 60% ACN, 0.1% FA. Finally, the organic solvent was removed in a SpeedVac concentrator prior to LC-MS/MS analysis.
2.7. Liquid Chromatography (LC) and Mass Spectrometry (MS) Analysis
Dried peptide samples were reconstituted with 2% ACN-0.1% FA and quantified by NanoDropTM spectrophometer (Thermo Fisher Scientific, Waltham, MA, USA) prior to LC-MS/MS analysis using a Proxeon EASY nanoLC system coupled to a Q-Exactive Plus mass spectrometer (Thermo Fisher Scientific, Waltham, MA, USA). Peptides were separated using an analytical C18 Acclaim PepMap column (75 µm × 250 mm, 2 µm particles; Thermo Fisher Scientific, Waltham, MA, USA) at a flow rate of 300 µL/min using a 118-min gradient: 1% to 6% B in 1 min, 6% to 23% B in 72 min, and 23% to 34% B in 45 min (A = FA, 0.1%; B = 80% ACN: 0.1% FA). The mass spectrometer was operated in positive data-dependent acquisition mode. MS1 spectra were measured with a resolution of 70,000 (AGC target: 1e6; mass range: 350–1700 m/z). Up to 12 MS2 spectra per duty cycle were triggered, fragmented by HCD, and acquired with a resolution of 17,500 (AGC target 5e4, isolation window; 1.2 m/z; normalized collision: 32). Dynamic exclusion was enabled with a duration of 25 s.
2.8. Data Analysis
All mass spectra from were analyzed with MaxQuant software version 1.5.5.1. MS/MS spectra were searched against the
Homo sapiens Uniprot protein sequence database (version January 2018) and GPM cRAP sequences (commonly known protein contaminants). Precursor mass tolerance was set to 20 ppm and 4.5 ppm for the first search where initial mass recalibration was completed and for the main search, respectively. Product ions were searched with a mass tolerance 0.5 Da. The maximum precursor ion charge state used for searching was 7. Carbamidomethylation of cysteines was searched as a fixed modification, while oxidation of methionines and acetylation of protein N-terminal were searched as variable modifications. Enzyme was set to trypsin in a specific mode and a maximum of two missed cleavages was allowed for searching. The target-decoy-based false discovery rate (FDR) filter for spectrum and protein identification was set to 1%. Proteins were classified as candidate interactors if they were identified in all three triplicate samples and abundances were at least 10-fold greater compared to respective controls. The STRING database (
www.string-db.org) was utilized for visualizing protein interaction clusters and cellular component GO enrichment analysis. The Retrieve/ID Mapping tool was utilized at
www.UniProt.org for subcellular location designations of identified candidate proteins.
4. Discussion
Our studies in live cells stably expressing TurboID and miniTurboID further support prior observation of potential toxicity and/or protein instability issues involving TurboID and miniTurbo that were reported in
C. elegans and
D. melanogaster [
34]. Cells expressing miniTurbo, and to a lesser extent TurboID, exhibited reduced levels of the ligase compared to BioID, possibly due to selection against cells with elevated expression and/or rapid degradation of these ligases. These toxicity and/or instability issues led us to exclude miniTurbo from subsequent studies as conventional PPI experiments in unhealthy cells could confound results. The cellular toxicity of these TurboID and especially miniTurboID proteins could come from persistent biotinylation of various proteins leading to their dysfunction. Another more likely source of toxicity is that these ligases could be competing with the endogenous biotin ligase for access to free biotin necessary to biotinylate key metabolic enzymes, a finding supported by the need for biotin supplementation to enable survival of a fly model stably expressing TurboID [
34]. Together, this new data suggests investigators should be cautious about using miniTurbo for proximity-labeling studies.
Our initial data comparing BioID- and TurboID-fused proteins revealed persistent steady-state biotinylation by TurboID under normal cell culture conditions that confounds its use for comparative studies using short labeling periods. In all of our experiments, TurboID persistently biotinylated proximal proteins at levels similar to a conventional induced BioID experiment. These findings emphasize the importance of validating the inducibility of biotinylation in TurboID-expressing cells prior to proximity-labeling experiments to ensure that there is nominal pre-existing biotinylation, and, when relevant, would support the use of no-biotin controls for proteomic experiments to ensure that detected proteins are unique to the exogenous biotin-induced labeling period.
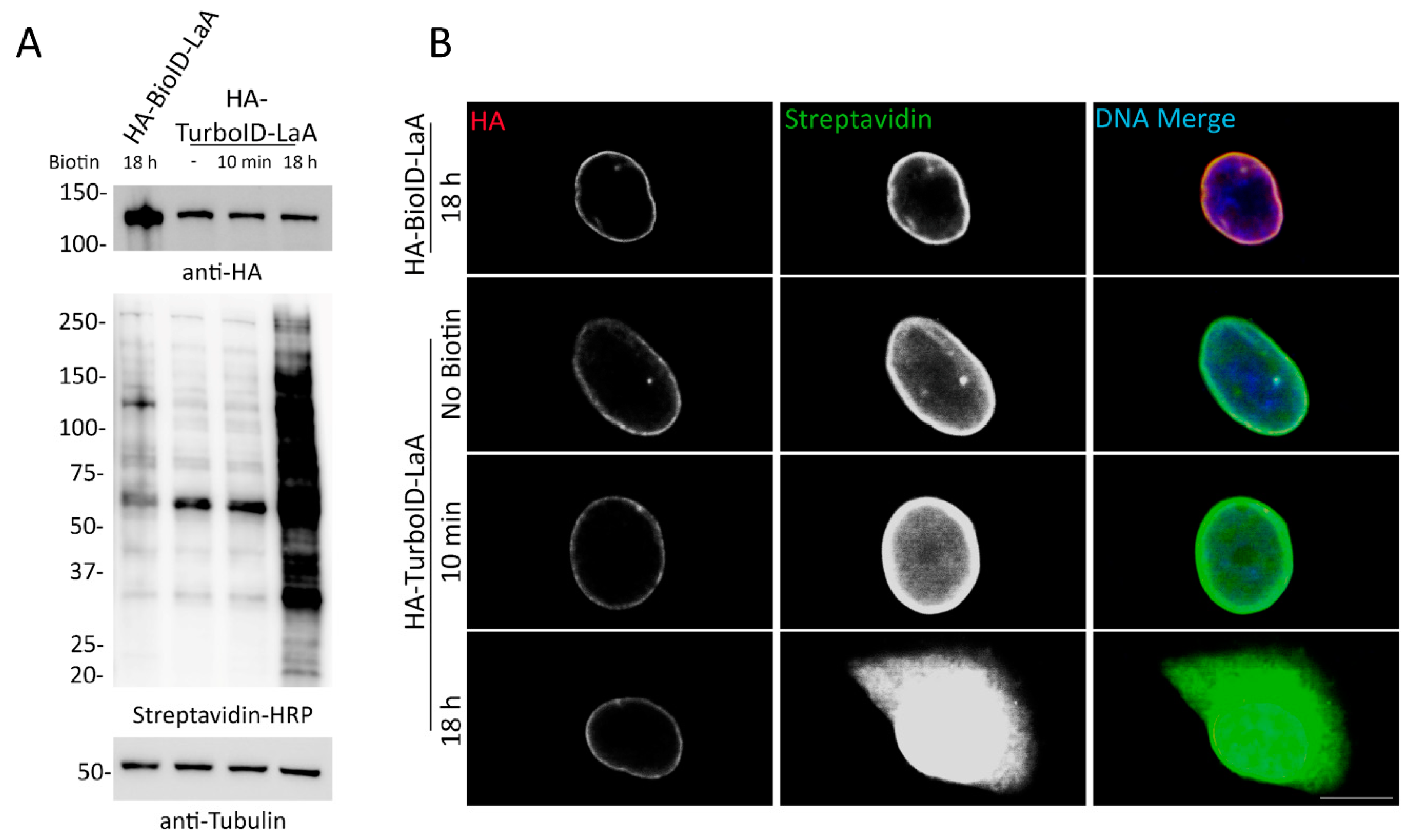
The total number of proteins identified by TurboID-LaA compared to BioID-LaA likely reflects an increased practical labeling radius and could prove problematic when attempting to map candidate PPI networks. We use the term ‘practical’ to reflect the distribution of the candidates identified by MS following a BioID or TurboID pulldown. The labeling radius may actually be increased for TurboID due to the saturation of available biotinylation sites in close proximity to the ligase that could promote labeling of sites further away. Alternatively, this may also reflect that the ligase is always active, albeit at lower levels in a non-biotin supplemented condition, and subpopulations of the bait proteins are in fact associated with these candidates throughout various stages of the cell cycle, perhaps especially during mitosis when there is no nuclear envelope and the nuclear lamina is disassembled. Another possible reason for some of the discrepancy in some of the candidates between BioID-LaA and TurboID-LaA may reflect a genuine difference in association of these proteins due to the cellular toxicity observed with TurboID constructs. Regardless of the reasons, the increase in proteins designated endoplasmic reticulum, cytoplasm, mitochondrion, and all other subcellular locations in the TurboID-identified candidates compared to the BioID-identified candidates forecasts a daunting task of further deciphering which proteins are relevant interactors versus irrelevant background proteins. What is clear however, is that the more abundant proteins detected by TurboID-LaA are most similar to those for BioID-LaA and are more likely to reflect proximate protein associations. The less abundant proteins are the ones that are more disparate.
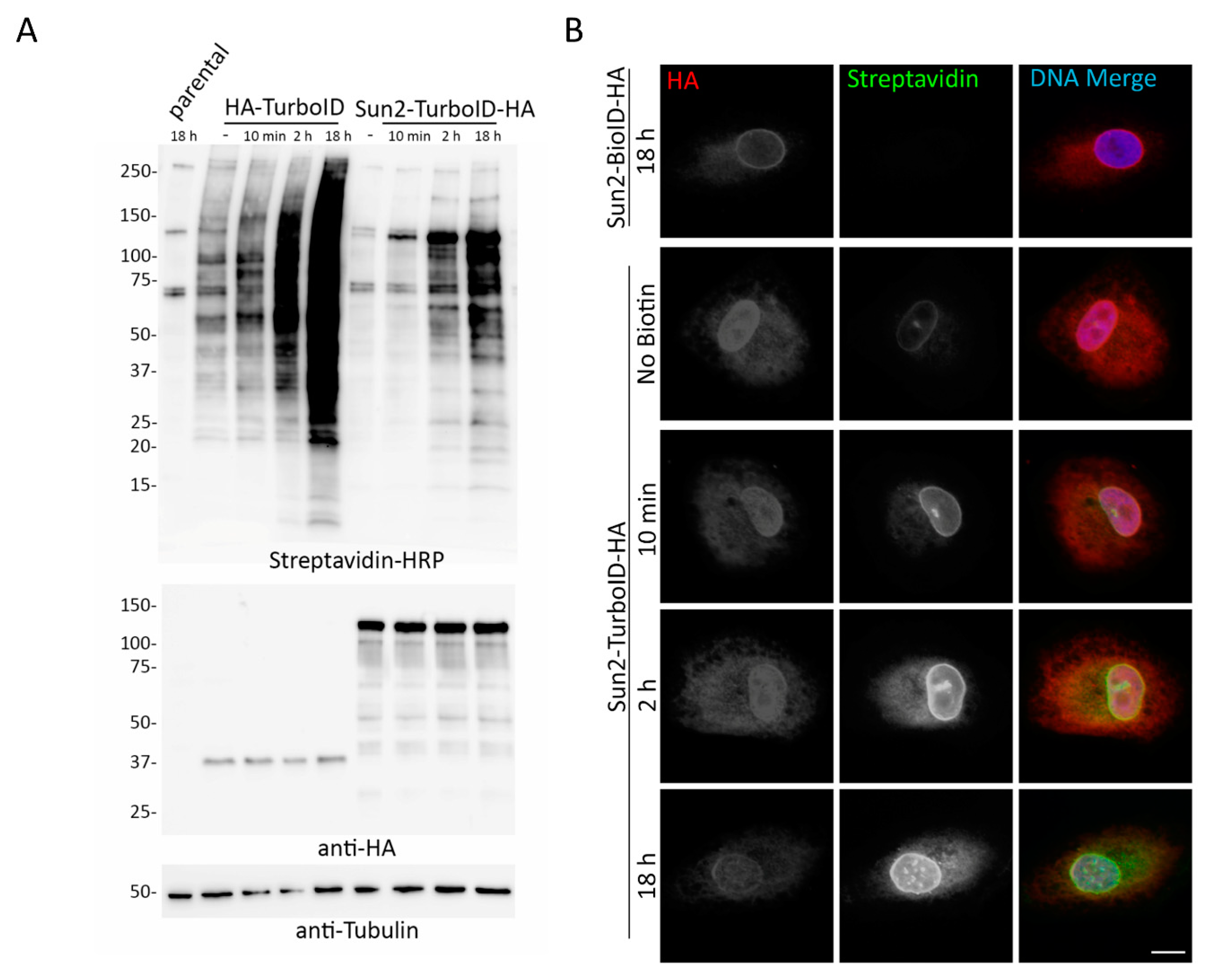
Our application of TurboID to Nups in the NPC also supported these inducibility of biotinylation and labeling radius issues. Proteomic analysis of TurboID-Nup53 cells at different timepoints following supplementation with biotin and isolation of biotinylated proteins demonstrates that TurboID does indeed label known proximate proteins without the addition of biotin and that further biotin supplementation only further increases the practical labeling radius of TurboID. For this reason, cells constitutively expressing TurboID will be uninducible for biotinylation in the conventional sense—that is they will continuously biotinylate proximate proteins under traditional culture conditions, which may preclude use of this faster ligase from certain experiments requiring short labeling periods, one of the primary motivations for the creation of TurboID.
In order to develop a new protocol that enabled reliable PPI-detection without the data-contamination that would occur as a result of perpetual biotinylation, we assessed several intermediate TurboID variants, tested transient transfection, utilized dox-inducible expression of TurboID, and cultured cells in dialyzed FBS that lacks biotin. We found that of these methods tested, combining inducible expression of TurboID with the use of dialyzed FBS allowed for inducible biotinylation by TurboID. Investigators who seek to compare a single cell line under multiple conditions should consider utilizing this approach to allow for detection of subtle changes or transient interactions that may otherwise be undetectable in a cell with pre-existing biotinylation of proximate proteins. Due to the increasing practical labeling radius that occurs with the length of biotin supplementation, it may be necessary for investigators to perform preliminary time-course experiments to determine the optimal length of biotin incubation as our studies show varying degrees of biotinylation for each bait tested.
Overall, these studies support TurboID as a rapid, promiscuous biotin ligase that can utilize basal levels of biotin in cell culture media for producing reactive bioAMP molecules. TurboID likely shares a similar mechanism of action with BioID, where both ligases create and release these reactive bioAMP molecules, but TurboID clearly does so at a much faster rate likely due, at least in part, to an enhanced affinity to biotin [
9,
10]. The expanding practical labeling radius evident with increasing biotin concentration and incubation duration may therefore impair the use of TurboID for some common BioID applications, but does not prohibit the use of TurboID under all circumstances. If the investigator is solely interested in elucidating the PPIs of a single bait under a single condition, it may be possible to utilize TurboID culture under normal media conditions and lyse the cells for pull down without supplementing with biotin. Additionally, if a BioID-bait fusion protein expression is extremely low due to toxicity, degradation, or suppression, it may be beneficial to try TurboID as the robust levels of biotinylation should be able to overcome low levels of expression. Furthermore, our studies support TurboID as the clearly superior ligase in the ER lumen with the ability to robustly biotinylate proteins in an environment where BioID is only marginally effective. Perhaps TurboID will be useful for other environments that may affect conventional BioID function such as organelles with low pH or extracellularly. Subsequent studies exploring these possibilities will be necessary to determine just how effectively TurboID will operate in these environments. It is clear that there is still the need for a proximity labeling method for living cells that has a well-defined practical labeling radius and that can be rapidly induced to enable studies of brief temporally distinct differences in protein associations. Whether this comes in the form of further modifications of labeling protocols with the existing ligases, modification to existing ligases, and/or entirely different approaches to proximity protein labeling remains to be seen.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}









