Signaling of Macrophages that Contours the Tumor Microenvironment for Promoting Cancer Development

Abstract
:1. Introduction
2. Macrophage Subtypes after Polarization and Signaling that Leads to their Polarization
3. Macrophage Subtypes and their Involvement in Cancer Initiation and Development through their Signaling to Re-Shape the Tumor Environment
3.1. The Cell Signaling and Subsequent Activation of Transcription Factors in Tumor-associated Macrophage (TAM) that Regulate Cancer Initiation
3.2. The Cell Signaling and Regulated Transcription Factors of TAM that Promote Cancer Progression
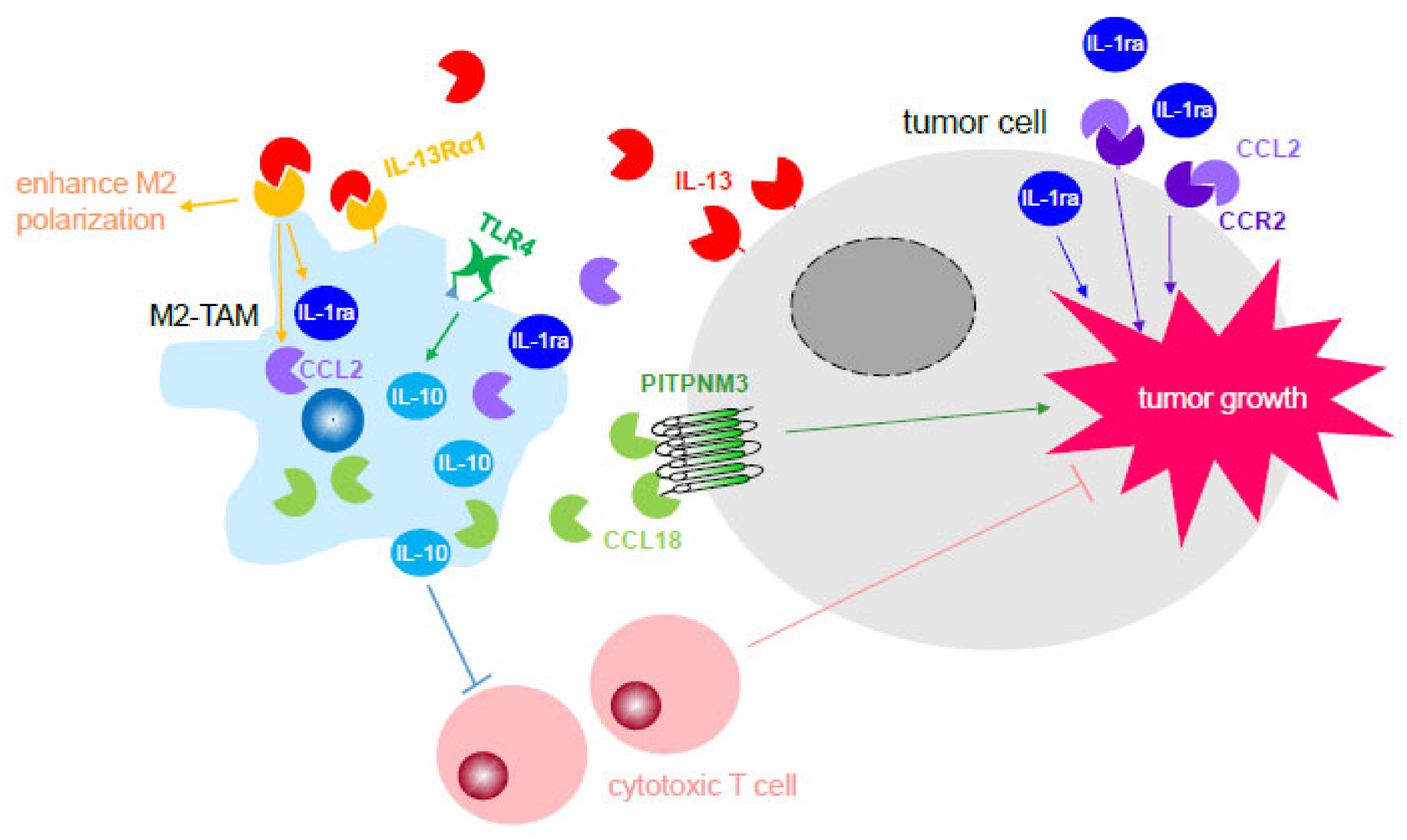
3.2.1. Activation of Cell-stimulating Growth Factors and Cytokines through Receptor Recognition to Directly Promote Tumor Progression
3.2.2. Suppression of Anti-tumor Immunity to Indirectly Support Cancer Progression
3.3. The Cell Signaling and its Regulated Transcription Factors of TAM that Control Cancer Metastasis

{kind=link}
{kind=link}
| TAM | Cancer | Reference | ||
|---|---|---|---|---|
| Cytokine | Signaling Pathway | Signaling Target | Type | |
| Wnt 5a | JNK | Ap-1/c-Jun, MMP7 | breast cancer | [70] |
| CXCL1 | NF-κB | NF-κB, Sox4 | breast cancer | [71] |
| intergrin α4 | PI3K, Akt | ND * | breast cancer | [72] |
| MMP9 | PI3K, Akt | snail | gastric cancer | [74] |
| COX-2 | Akt | ND * | breast cancer | [75] |
| IL-10 | ND | snail, vimentin | pancreatic cancer | [41] |
| MMP9 | ND | ND * | larynx carcinoma | [79] |
| TGFβ1 | ND | MMP9 | glioma | [80] |
| EGF | ERK, IncRNA | N-cadherin, vimentin | ovarian cancer | [81] |
| ND ** | ERK | slug | lung cancer | [82,83] |
4. Conclusion Remarks
Funding
Conflicts of Interest
Abbreviations
| ADM | acinar-to-ductal metaplasia |
| AR | androgen receptor |
| CSF-1 | colony-stimulating factor 1 |
| FPR2 | formyl peptide receptor 2 |
| GDNF | glial cell-derived neurotrophic factor |
| GPR18 | G protein-coupled receptor 18 |
| ECM | extracellular matrix |
| EGF | epithelial growth factor |
| EMR1 | EGF-like module-containing mucin-like hormone receptor-like 1 |
| EMT | epithelial–mesenchymal transition |
| ERK | extracellular signal-regulated kinase |
| GSK3β | glycogen synthase kinase 3β |
| HCC | hepatocellular carcinoma |
| ICAM-1 | intracellular adhesion molecule 1 |
| IFN-gamma | interferon gamma |
| IL-1 ra | interleukin 1 antagonist |
| IL-6 | interleukin 6 |
| JAK | Janus kinase |
| LIMT | IncRNA inhibiting metastasis |
| LPS | lipopolysaccharide |
| MCP-1 | monocyte chemoattractant protein 1 |
| MHC | major histocompatibility complex |
| MMP | matrix metalloprotease |
| NF-κB | nuclear factor kappa-light-chain-enhancer if activated B cells |
| NK | natural killer |
| PanIN | pancreatic intraepithelial neoplasia |
| PDAC | pancreatic ductal adenocarcinoma |
| PI3K | phosphatidylinositol 3-kinase |
| PIPNM3 | phosphatidylinositol transfer protein membrane-associated 3 |
| PLGF | placental growth factor |
| PTEN | phosphatase and tensin homolog |
| RANTES | regulated on activation, normal T cell expressed and secreted |
| STAT3 | signal transducer and activator of transcription 3 |
| TAM | tumor-associated macrophage |
| Treg | regulatory T |
| TGF-β | transforming growth factor β |
| TLR | toll-like receptor |
| TNF | tumor necrosis factor |
| VCAM-1 | vascular cell adhesion protein 1 |
| VEGF | vascular endothelial growth factor |
| VEGFR | vascular endothelial growth factor receptor |
| ZEB1 | zinc finger E-box-binding homeobox 1 |
References
- Arango Duque, G.; Descoteaux, A. Macrophage cytokines: Involvement in immunity and infectious diseases. Front. Immunol. 2014, 5, 491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Navegantes, K.C.; de Souza Gomes, R.; Pereira, P.A.T.; Czaikoski, P.G.; Azevedo, C.H.M.; Monteiro, M.C. Immune modulation of some autoimmune diseases: The critical role of macrophages and neutrophils in the innate and adaptive immunity. J. Transl. Med. 2017, 15, 36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parihar, A.; Eubank, T.D.; Doseff, A.I. Monocytes and macrophages regulate immunity through dynamic networks of survival and cell death. J. Innate Immun. 2010, 2, 204–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turner, M.D.; Nedjai, B.; Hurst, T.; Pennington, D.J. Cytokines and chemokines: At the crossroads of cell signalling and inflammatory disease. Biochim. Biophys. Acta. 2014, 1843, 2563–2582. [Google Scholar] [CrossRef] [Green Version]
- Aras, S.; Zaidi, M.R. TAMeless traitors: Macrophages in cancer progression and metastasis. Br. J. Cancer 2017, 117, 1583–1591. [Google Scholar] [CrossRef] [Green Version]
- Jayasingam, S.D.; Citartan, M.; Thang, T.H.; Mat Zin, A.A.; Ang, K.C.; Ch’ng, E.S. Evaluating the Polarization of Tumor-Associated Macrophages Into M1 and M2 Phenotypes in Human Cancer Tissue: Technicalities and Challenges in Routine Clinical Practice. Front. Oncol 2019, 9, 1512. [Google Scholar] [CrossRef] [Green Version]
- Suarez-Carmona, M.; Lesage, J.; Cataldo, D.; Gilles, C. EMT and inflammation: Inseparable actors of cancer progression. Mol. Oncol. 2017, 11, 805–823. [Google Scholar] [CrossRef]
- Takeya, M.; Komohara, Y. Role of tumor-associated macrophages in human malignancies: Friend or foe? Pathol. Int. 2016, 66, 491–505. [Google Scholar] [CrossRef]
- Yahaya, M.A.F.; Lila, M.A.M.; Ismail, S.; Zainol, M.; Afizan, N. Tumour-Associated Macrophages (TAMs) in Colon Cancer and How to Reeducate Them. J. Immunol. Res. 2019, 2019, 2368249. [Google Scholar] [CrossRef]
- Yin, M.; Shen, J.; Yu, S.; Fei, J.; Zhu, X.; Zhao, J.; Zhai, L.; Sadhukhan, A.; Zhou, J. Tumor-Associated Macrophages (TAMs): A Critical Activator In Ovarian Cancer Metastasis. Onco. Targets. Ther. 2019, 12, 8687–8699. [Google Scholar] [CrossRef] [Green Version]
- Arnold, C.E.; Whyte, C.S.; Gordon, P.; Barker, R.N.; Rees, A.J.; Wilson, H.M. A critical role for suppressor of cytokine signalling 3 in promoting M1 macrophage activation and function in vitro and in vivo. Immunology 2014, 141, 96–110. [Google Scholar] [CrossRef] [Green Version]
- MacMicking, J.; Xie, Q.W.; Nathan, C. Nitric oxide and macrophage function. Annu. Rev. Immunol. 1997, 15, 323–350. [Google Scholar] [CrossRef] [PubMed]
- Mills, C.D. M1 and M2 Macrophages: Oracles of Health and Disease. Crit Rev. Immunol. 2012, 32, 463–488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muraille, E.; Leo, O.; Moser, M. TH1/TH2 paradigm extended: Macrophage polarization as an unappreciated pathogen-driven escape mechanism? Front. Immunol. 2014, 5, 603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Choksi, S.; Chen, K.; Pobezinskaya, Y.; Linnoila, I.; Liu, Z.G. ROS play a critical role in the differentiation of alternatively activated macrophages and the occurrence of tumor-associated macrophages. Cell. Res. 2013, 23, 898–914. [Google Scholar] [CrossRef] [Green Version]
- Jablonski, K.A.; Amici, S.A.; Webb, L.M.; Ruiz-Rosado Jde, D.; Popovich, P.G.; Partida-Sanchez, S.; Guerau-de-Arellano, M. Novel markers to delineate murine M1 and M2 macrophages. PLoS. ONE. 2015, 10. [Google Scholar] [CrossRef] [Green Version]
- Sheikh, F.; Dickensheets, H.; Pedras-Vasconcelos, J.; Ramalingam, T.; Helming, L.; Gordon, S.; Donnelly, R.P. The Interleukin-13 Receptor-alpha1 Chain Is Essential for Induction of the Alternative Macrophage Activation Pathway by IL-13 but Not IL-4. J. Innate. Immun. 2015, 7, 494–505. [Google Scholar] [CrossRef] [Green Version]
- Sica, A.; Mantovani, A. Macrophage plasticity and polarization: In Vivo veritas. J. Clin. Invest. 2012, 122, 787–795. [Google Scholar] [CrossRef]
- Rocher, C.; Singla, D.K. SMAD-PI3K-Akt-mTOR pathway mediates BMP-7 polarization of monocytes into M2 macrophages. PLoS. ONE 2013, 8, e84009. [Google Scholar] [CrossRef] [Green Version]
- Lawrence, T.; Natoli, G. Transcriptional regulation of macrophage polarization: Enabling diversity with identity. Nat. Rev. Immunol 2011, 11, 750–761. [Google Scholar] [CrossRef]
- Mantovani, A.; Sozzani, S.; Locati, M.; Allavena, P.; Sica, A. Macrophage polarization: Tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends. Immunol. 2002, 23, 549–555. [Google Scholar] [CrossRef]
- Little, A.C.; Pathanjeli, P.; Wu, Z.; Bao, L.; Goo, L.E.; Yates, J.A.; Oliver, C.R.; Soellner, M.B.; Merajver, S.D. IL-4/IL-13 Stimulated Macrophages Enhance Breast Cancer Invasion Via Rho-GTPase Regulation of Synergistic VEGF/CCL-18 Signaling. Front. Oncol. 2019, 9, 456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allavena, P.; Sica, A.; Solinas, G.; Porta, C.; Mantovani, A. The inflammatory micro-environment in tumor progression: The role of tumor-associated macrophages. Crit Rev. Oncol. Hematol. 2008, 66, 1–9. [Google Scholar] [CrossRef]
- Bai, J.; Adriani, G.; Dang, T.M.; Tu, T.Y.; Penny, H.X.; Wong, S.C.; Kamm, R.D.; Thiery, J.P. Contact-dependent carcinoma aggregate dispersion by M2a macrophages via ICAM-1 and beta2 integrin interactions. Oncotarget. 2015, 6, 25295–25307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sousa, S.; Brion, R.; Lintunen, M.; Kronqvist, P.; Sandholm, J.; Monkkonen, J.; Kellokumpu-Lehtinen, P.L.; Lauttia, S.; Tynninen, O.; Joensuu, H.; et al. Human breast cancer cells educate macrophages toward the M2 activation status. Breast Cancer Res. 2015, 17, 101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsieh, C.S.; Macatonia, S.E.; O’Garra, A.; Murphy, K.M. Pathogen-induced Th1 phenotype development in CD4+ alpha beta-TCR transgenic T cells is macrophage dependent. Int. Immunol. 1993, 5, 371–382. [Google Scholar] [CrossRef]
- Swain, S.L.; Weinberg, A.D.; English, M.; Huston, G. IL-4 directs the development of Th2-like helper effectors. J. Immunol. 1990, 145, 3796–3806. [Google Scholar]
- Linterman, M.A.; Vinuesa, C.G. T follicular helper cells during immunity and tolerance. Prog. Mol. Biol. Transl. Sci. 2010, 92, 207–248. [Google Scholar] [CrossRef]
- Alao, J.P. The regulation of cyclin D1 degradation: Roles in cancer development and the potential for therapeutic invention. Mol. Cancer. 2007, 6, 24. [Google Scholar] [CrossRef] [Green Version]
- Kong, L.; Zhou, Y.; Bu, H.; Lv, T.; Shi, Y.; Yang, J. Deletion of interleukin-6 in monocytes/macrophages suppresses the initiation of hepatocellular carcinoma in mice. J. Exp. Clin. Cancer Res. 2016, 35, 131. [Google Scholar] [CrossRef] [Green Version]
- Soresi, M.; Giannitrapani, L.; D’Antona, F.; Florena, A.M.; La Spada, E.; Terranova, A.; Cervello, M.; D’Alessandro, N.; Montalto, G. Interleukin-6 and its soluble receptor in patients with liver cirrhosis and hepatocellular carcinoma. World. J. Gastroenterol. 2006, 12, 2563–2568. [Google Scholar] [CrossRef]
- Liou, G.Y.; Doppler, H.; Necela, B.; Krishna, M.; Crawford, H.C.; Raimondo, M.; Storz, P. Macrophage-secreted cytokines drive pancreatic acinar-to-ductal metaplasia through NF-kappaB and MMPs. J. Cell. Biol. 2013, 202, 563–577. [Google Scholar] [CrossRef] [PubMed]
- Liou, G.Y.; Doppler, H.; Necela, B.; Edenfield, B.; Zhang, L.; Dawson, D.W.; Storz, P. Mutant KRAS-induced expression of ICAM-1 in pancreatic acinar cells causes attraction of macrophages to expedite the formation of precancerous lesions. Cancer Discov. 2015, 5, 52–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graeber, M.B.; Scheithauer, B.W.; Kreutzberg, G.W. Microglia in brain tumors. Glia. 2002, 40, 252–259. [Google Scholar] [CrossRef] [PubMed]
- Glass, R.; Synowitz, M. CNS macrophages and peripheral myeloid cells in brain tumours. Acta. Neuropathol. 2014, 128, 347–362. [Google Scholar] [CrossRef] [Green Version]
- Chia, K.; Mazzolini, J.; Mione, M.; Sieger, D. Tumor initiating cells induce Cxcr4-mediated infiltration of pro-tumoral macrophages into the brain. Elife. 2018, 7. [Google Scholar] [CrossRef]
- Dang, T.; Liou, G.Y. Macrophage Cytokines Enhance Cell Proliferation of Normal Prostate Epithelial Cells through Activation of ERK and Akt. Sci. Rep. 2018, 8, 7718. [Google Scholar] [CrossRef]
- Epelman, S.; Lavine, K.J.; Randolph, G.J. Origin and functions of tissue macrophages. Immunity 2014, 41, 21–35. [Google Scholar] [CrossRef] [Green Version]
- Fang, L.Y.; Izumi, K.; Lai, K.P.; Liang, L.; Li, L.; Miyamoto, H.; Lin, W.J.; Chang, C. Infiltrating macrophages promote prostate tumorigenesis via modulating androgen receptor-mediated CCL4-STAT3 signaling. Cancer Res. 2013, 73, 5633–5646. [Google Scholar] [CrossRef] [Green Version]
- Mano, Y.; Aishima, S.; Fujita, N.; Tanaka, Y.; Kubo, Y.; Motomura, T.; Taketomi, A.; Shirabe, K.; Maehara, Y.; Oda, Y. Tumor-associated macrophage promotes tumor progression via STAT3 signaling in hepatocellular carcinoma. Pathobiology 2013, 80, 146–154. [Google Scholar] [CrossRef]
- Liu, C.Y.; Xu, J.Y.; Shi, X.Y.; Huang, W.; Ruan, T.Y.; Xie, P.; Ding, J.L. M2-polarized tumor-associated macrophages promoted epithelial-mesenchymal transition in pancreatic cancer cells, partially through TLR4/IL-10 signaling pathway. Lab. Invest. 2013, 93, 844–854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, Y.F.; Tan, Y.X.; Wang, M.; Zhang, J.; Zhang, B.; Yang, C.; Ding, Z.W.; Dong, L.W.; Wang, H.Y. Signal regulatory protein alpha is associated with tumor-polarized macrophages phenotype switch and plays a pivotal role in tumor progression. Hepatology 2013, 58, 680–691. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, Y.; Komohara, Y.; Ikeda, T.; Takeya, M. Corosolic acid inhibits glioblastoma cell proliferation by suppressing the activation of signal transducer and activator of transcription-3 and nuclear factor-kappa B in tumor cells and tumor-associated macrophages. Cancer Sci. 2011, 102, 206–211. [Google Scholar] [CrossRef] [PubMed]
- Liou, G.Y.; Bastea, L.; Fleming, A.; Doppler, H.; Edenfield, B.H.; Dawson, D.W.; Zhang, L.; Bardeesy, N.; Storz, P. The Presence of Interleukin-13 at Pancreatic ADM/PanIN Lesions Alters Macrophage Populations and Mediates Pancreatic Tumorigenesis. Cell. Rep. 2017, 19, 1322–1333. [Google Scholar] [CrossRef] [Green Version]
- Ye, H.; Zhou, Q.; Zheng, S.; Li, G.; Lin, Q.; Wei, L.; Fu, Z.; Zhang, B.; Liu, Y.; Li, Z.; et al. Tumor-associated macrophages promote progression and the Warburg effect via CCL18/NF-kB/VCAM-1 pathway in pancreatic ductal adenocarcinoma. Cell Death Dis. 2018, 9, 453. [Google Scholar] [CrossRef] [Green Version]
- Kaler, P.; Augenlicht, L.; Klampfer, L. Macrophage-derived IL-1beta stimulates Wnt signaling and growth of colon cancer cells: A crosstalk interrupted by vitamin D3. Oncogene 2009, 28, 3892–3902. [Google Scholar] [CrossRef] [Green Version]
- Kaler, P.; Godasi, B.N.; Augenlicht, L.; Klampfer, L. The NF-kappaB/AKT-dependent Induction of Wnt Signaling in Colon Cancer Cells by Macrophages and IL-1beta. Cancer Microenviron. 2009, 2, 69–80. [Google Scholar] [CrossRef] [Green Version]
- Takaishi, K.; Komohara, Y.; Tashiro, H.; Ohtake, H.; Nakagawa, T.; Katabuchi, H.; Takeya, M. Involvement of M2-polarized macrophages in the ascites from advanced epithelial ovarian carcinoma in tumor progression via Stat3 activation. Cancer Sci. 2010, 101, 2128–2136. [Google Scholar] [CrossRef]
- Zhang, X.; Zeng, Y.; Qu, Q.; Zhu, J.; Liu, Z.; Ning, W.; Zeng, H.; Zhang, N.; Du, W.; Chen, C.; et al. PD-L1 induced by IFN-gamma from tumor-associated macrophages via the JAK/STAT3 and PI3K/AKT signaling pathways promoted progression of lung cancer. Int. J. Clin. Oncol. 2017, 22, 1026–1033. [Google Scholar] [CrossRef]
- Wan, S.; Zhao, E.; Kryczek, I.; Vatan, L.; Sadovskaya, A.; Ludema, G.; Simeone, D.M.; Zou, W.; Welling, T.H. Tumor-associated macrophages produce interleukin 6 and signal via STAT3 to promote expansion of human hepatocellular carcinoma stem cells. Gastroenterology 2014, 147, 1393–1404. [Google Scholar] [CrossRef] [Green Version]
- Papaccio, F.; Paino, F.; Regad, T.; Papaccio, G.; Desiderio, V.; Tirino, V. Concise Review: Cancer Cells, Cancer Stem Cells, and Mesenchymal Stem Cells: Influence in Cancer Development. Stem Cells Transl. Med. 2017, 6, 2115–2125. [Google Scholar] [CrossRef]
- Gao, L.; Zhang, W.; Zhong, W.Q.; Liu, Z.J.; Li, H.M.; Yu, Z.L.; Zhao, Y.F. Tumor associated macrophages induce epithelial to mesenchymal transition via the EGFR/ERK1/2 pathway in head and neck squamous cell carcinoma. Oncol. Rep. 2018, 40, 2558–2572. [Google Scholar] [CrossRef]
- Cortes, M.; Sanchez-Moral, L.; de Barrios, O.; Fernandez-Acenero, M.J.; Martinez-Campanario, M.C.; Esteve-Codina, A.; Darling, D.S.; Gyorffy, B.; Lawrence, T.; Dean, D.C.; et al. Tumor-associated macrophages (TAMs) depend on ZEB1 for their cancer-promoting roles. EMBO J. 2017, 36, 3336–3355. [Google Scholar] [CrossRef]
- Cai, J.; Xia, L.; Li, J.; Ni, S.; Song, H.; Wu, X. Tumor-Associated Macrophages Derived TGF-betaInduced Epithelial to Mesenchymal Transition in Colorectal Cancer Cells through Smad2,3-4/Snail Signaling Pathway. Cancer Res. Treat. 2019, 51, 252–266. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Che, D.; Yang, F.; Chi, C.; Meng, H.; Shen, J.; Qi, L.; Liu, F.; Lv, L.; Li, Y.; et al. Tumor-associated macrophages promote tumor metastasis via the TGF-beta/SOX9 axis in non-small cell lung cancer. Oncotarget. 2017, 8, 99801–99815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dos Reis, D.C.; Damasceno, K.A.; de Campos, C.B.; Veloso, E.S.; Pegas, G.R.A.; Kraemer, L.R.; Rodrigues, M.A.; Mattos, M.S.; Gomes, D.A.; Campos, P.P.; et al. Versican and Tumor-Associated Macrophages Promotes Tumor Progression and Metastasis in Canine and Murine Models of Breast Carcinoma. Front. Oncol 2019, 9, 577. [Google Scholar] [CrossRef] [PubMed]
- Qiu, S.; Deng, L.; Liao, X.; Nie, L.; Qi, F.; Jin, K.; Tu, X.; Zheng, X.; Li, J.; Liu, L.; et al. Tumor-associated macrophages promote bladder tumor growth through PI3K/AKT signal induced by collagen. Cancer Sci. 2019, 110, 2110–2118. [Google Scholar] [CrossRef] [PubMed]
- Luna, J.I.; Grossenbacher, S.K.; Murphy, W.J.; Canter, R.J. Targeting cancer stem cells with natural killer cell immunotherapy. Expert Opin. Biol. Ther. 2017, 17, 313–324. [Google Scholar] [CrossRef] [Green Version]
- Kusmartsev, S.; Gabrilovich, D.I. STAT1 signaling regulates tumor-associated macrophage-mediated T cell deletion. J. Immunol. 2005, 174, 12. [Google Scholar] [CrossRef] [Green Version]
- Bolpetti, A.; Silva, J.S.; Villa, L.L.; Lepique, A.P. Interleukin-10 production by tumor infiltrating macrophages plays a role in Human Papillomavirus 16 tumor growth. BMC Immunol. 2010, 11. [Google Scholar] [CrossRef] [Green Version]
- Dahmani, A.; Delisle, J.S. TGF-β in T cell biology: Implications for cancer immunotherapy. Cancers (Basel) 2018, 10, 194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Z.; Kuang, W.; Zhou, Q.; Zhang, Y. TGF-β1 secreted by M2 phenotype macrophages enhances the stemness and migration of glioma cells via the SMAD2/3 signalling pathway. Int. J. Mol. Med. 2018, 42, 3395–3403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Liu, J.Q.; Talebian, F.; Liu, Z.; Yu, L.; Bai, X.F. IL-10 enhances CTL-mediated tumor rejection by inhibiting highly suppressive CD4+ T cells and promoting CTL persistence in a murine model of plasmacytoma. Oncoimmunology 2015, 4, e1014232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arlauckas, S.P.; Garris, C.S.; Kohler, R.H.; Kitaoka, M.; Cuccarese, M.F.; Yang, K.S.; Miller, M.A.; Carlson, J.C.; Freeman, G.J.; Anthony, R.M.; et al. In vivo imaging reveals a tumor-associated macrophage-mediated resistance pathway in anti-PD-1 therapy. Sci. Transl Med. 2017, 9, eaal3604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Q.; Wu, X.; Wu, Y.; Wang, X. Interaction between Treg cells and tumor-associated macrophages in the tumor microenvironment of epithelial ovarian cancer. Oncol Rep. 2016, 36, 3472–3478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trinchieri, G. Interleukin-10 production by effector T cells: Th1 cells show self control. J. Exp. Med. 2007, 204, 239–243. [Google Scholar] [CrossRef]
- Lai, Y.S.; Wahyuningtyas, R.; Aui, S.P.; Chang, K.T. Autocrine VEGF signalling on M2 macrophages regulates PD-L1 expression for immunomodulation of T cells. J. Cell Mol. Med. 2019, 23, 1257–1267. [Google Scholar] [CrossRef]
- Henze, A.T.; Mazzone, M. The impact of hypoxia on tumor-associated macrophages. J. Clin. Invest. 2016, 126, 3672–3679. [Google Scholar] [CrossRef]
- Jeong, H.; Kim, S.; Hong, B.J.; Lee, C.J.; Kim, Y.E.; Bok, S.; Oh, J.M.; Gwak, S.H.; Yoo, M.Y.; Lee, M.S.; et al. Tumor-Associated Macrophages Enhance Tumor Hypoxia and Aerobic Glycolysis. Cancer Res. 2019, 79, 795–806. [Google Scholar] [CrossRef] [Green Version]
- Pukrop, T.; Klemm, F.; Hagemann, T.; Gradl, D.; Schulz, M.; Siemes, S.; Trumper, L.; Binder, C. Wnt 5a signaling is critical for macrophage-induced invasion of breast cancer cell lines. Proc. Natl Acad. Sci. USA 2006, 103, 5454–5459. [Google Scholar] [CrossRef] [Green Version]
- Wang, N.; Liu, W.; Zheng, Y.; Wang, S.; Yang, B.; Li, M.; Song, J.; Zhang, F.; Zhang, X.; Wang, Q.; et al. CXCL1 derived from tumor-associated macrophages promotes breast cancer metastasis via activating NF-kappaB/SOX4 signaling. Cell Death Dis. 2018, 9, 880. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Zhang, X.H.; Massague, J. Macrophage binding to receptor VCAM-1 transmits survival signals in breast cancer cells that invade the lungs. Cancer Cell 2011, 20, 538–549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qian, B.Z.; Zhang, H.; Li, J.; He, T.; Yeo, E.J.; Soong, D.Y.; Carragher, N.O.; Munro, A.; Chang, A.; Bresnick, A.R.; et al. FLT1 signaling in metastasis-associated macrophages activates an inflammatory signature that promotes breast cancer metastasis. J. Exp. Med. 2015, 212, 1433–1448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, L.; Ye, Y.; Zhu, X. MMP-9 secreted by tumor associated macrophages promoted gastric cancer metastasis through a PI3K/AKT/Snail pathway. Biomed. Pharmacother 2019, 117, 109096. [Google Scholar] [CrossRef]
- Gan, L.; Qiu, Z.; Huang, J.; Li, Y.; Huang, H.; Xiang, T.; Wan, J.; Hui, T.; Lin, Y.; Li, H.; et al. Cyclooxygenase-2 in tumor-associated macrophages promotes metastatic potential of breast cancer cells through Akt pathway. Int J. Biol. Sci. 2016, 12, 1533–1543. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez-Avila, G.; Sommer, B.; Mendoza-Posada, D.A.; Ramos, C.; Garcia-Hernandez, A.A.; Falfan-Valencia, R. Matrix metalloproteinases participation in the metastatic process and their diagnostic and therapeutic applications in cancer. Crit. Rev. Oncol. Hematol. 2019, 137, 57–83. [Google Scholar] [CrossRef]
- Shay, G.; Lynch, C.C.; Fingleton, B. Moving targets: Emerging roles for MMPs in cancer progression and metastasis. Matrix. Biol. 2015, 44-46, 200–206. [Google Scholar] [CrossRef]
- Kessenbrock, K.; Plaks, V.; Werb, Z. Matrix metalloproteinases: Regulators of the tumor microenvironment. Cell 2010, 141, 52–67. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.; Qi, Y. Larynx carcinoma regulates tumor-associated macrophages through PLGF signaling. Sci. Rep. 2015, 5, 10071. [Google Scholar] [CrossRef] [Green Version]
- Ye, X.Z.; Xu, S.L.; Xin, Y.H.; Yu, S.C.; Ping, Y.F.; Chen, L.; Xiao, H.L.; Wang, B.; Yi, L.; Wang, Q.L.; et al. Tumor-associated microglia/macrophages enhance the invasion of glioma stem-like cells via TGF-beta1 signaling pathway. J. Immunol. 2012, 189, 444–453. [Google Scholar] [CrossRef] [Green Version]
- Zeng, X.Y.; Xie, H.; Yuan, J.; Jiang, X.Y.; Yong, J.H.; Zeng, D.; Dou, Y.Y.; Xiao, S.S. M2-like tumor-associated macrophages-secreted EGF promotes epithelial ovarian cancer metastasis via activating EGFR-ERK signaling and suppressing lncRNA LIMT expression. Cancer Biol. Ther. 2019, 20, 956–966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, Z.; Song, J.; Hao, J.; Zhao, H.; Du, X.; Li, E.; Kuang, Y.; Yang, F.; Wang, W.; Deng, J.; et al. M2 macrophages promote NSCLC metastasis by upregulating CRYAB. Cell Death Dis. 2019, 10, 377. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Cao, J.; Ma, S.; Dong, R.; Meng, W.; Ying, M.; Weng, Q.; Chen, Z.; Ma, J.; Fang, Q.; et al. Tumor hypoxia enhances Non-Small Cell Lung Cancer metastasis by selectively promoting macrophage M2 polarization through the activation of ERK signaling. Oncotarget. 2014, 5, 9664–9677. [Google Scholar] [CrossRef] [PubMed] [Green Version]


| Macrophage Subtype. | M1 | M2a | M2b | M2c | M2d |
|---|---|---|---|---|---|
| Stimulation | IFNγ, LPS, GM-CSF | IL-4, IL-13, fungal infection | IL-1R | IL-10, TGFβ, glucocorticoids | IL-6, leukocyte inhibitory factor, adenosine |
| Receptor | IFNγR, TLR4, CSF2Rα | TLR4, IL-4Rα | TLR4, IL-4Rα | TLR4, IL-4Rα | TLR4, IL-4Rα |
| Signaling pathway | JAK1/2, P38, MSK1/2 | p38 MAPK, JAK1/2/3, PI3K | p38 MAPK, JAK1/2/3, PI3K | p38 MAPK, JAK1/2/3, PI3K | p38 MAPK, JAK1/2/3, PI3K |
| Transcription Factors | STAT1/2/5, AP1, IRF3/5, NF-κB | CREB, JMJD3, STAT6, IRF4, PPARγ | CREB, JMJD3, STAT6, IRF4, C/EBPβ | CREB, JMJD3, STAT6, IRF4, C/EBPβ | CREB, JMJD3, STAT6, IRF4, C/EBPβ |
| Target Genes | Nos2, Ciita, IL12b, inflammatory genes | Arg1, FIzz1, Ym1, CD206 | Arg1, Fizz1, Ym1, CD206 | Arg1, Fizz1, Ym1, CD206 | Arg1, Fizz1, Ym1, CD206 |
| Cytokine Secretion | TNF, IL-1, IL-6, IL-12, IL-23 | IL-10, TGFβ, IL-1ra | IL-1, IL-6, IL-10, TNF | IL-10, TGFβ | IL-10, IL-12, TNF, TGFβ |
| Chemokine Secretion. | CCL10, CCL11, CCL5, CCL8, CCL9 | CCL17, CCL22, CCL24 | CCL1 | CCR2 | CCL5, CXCL10, CXCL16 |
| TAM | Tumor | Reference | |
|---|---|---|---|
| Cytokine | Signaling Target | Type | |
| IL-6 * | STAT3, cyclin D1, c-myc | hepatocellular carcinoma | [30,31] |
| TNF, RANTES | NF-κB, MMP9 | pancreatic ductal adenocarcinoma | [32] |
| SDF1 | Akt, CXCR4 | gliomas | [36] |
| CCL3, IL-1ra, osteopontin, M- CSF1, GDNF | Akt, CXCR4 | prostate cancer | [37] |
| CCL4 * | STAT3, COX-2, c-myc, PTEN, p53 | prostate cancer | [39] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Messex, J.K.; Byrd, C.J.; Liou, G.-Y. Signaling of Macrophages that Contours the Tumor Microenvironment for Promoting Cancer Development. Cells 2020, 9, 919. https://doi.org/10.3390/cells9040919
Messex JK, Byrd CJ, Liou G-Y. Signaling of Macrophages that Contours the Tumor Microenvironment for Promoting Cancer Development. Cells. 2020; 9(4):919. https://doi.org/10.3390/cells9040919
Chicago/Turabian StyleMessex, Justin K., Crystal J. Byrd, and Geou-Yarh Liou. 2020. "Signaling of Macrophages that Contours the Tumor Microenvironment for Promoting Cancer Development" Cells 9, no. 4: 919. https://doi.org/10.3390/cells9040919