Diagnostic and Therapeutic Potential of TSPO Studies Regarding Neurodegenerative Diseases, Psychiatric Disorders, Alcohol Use Disorders, Traumatic Brain Injury, and Stroke: An Update


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Background
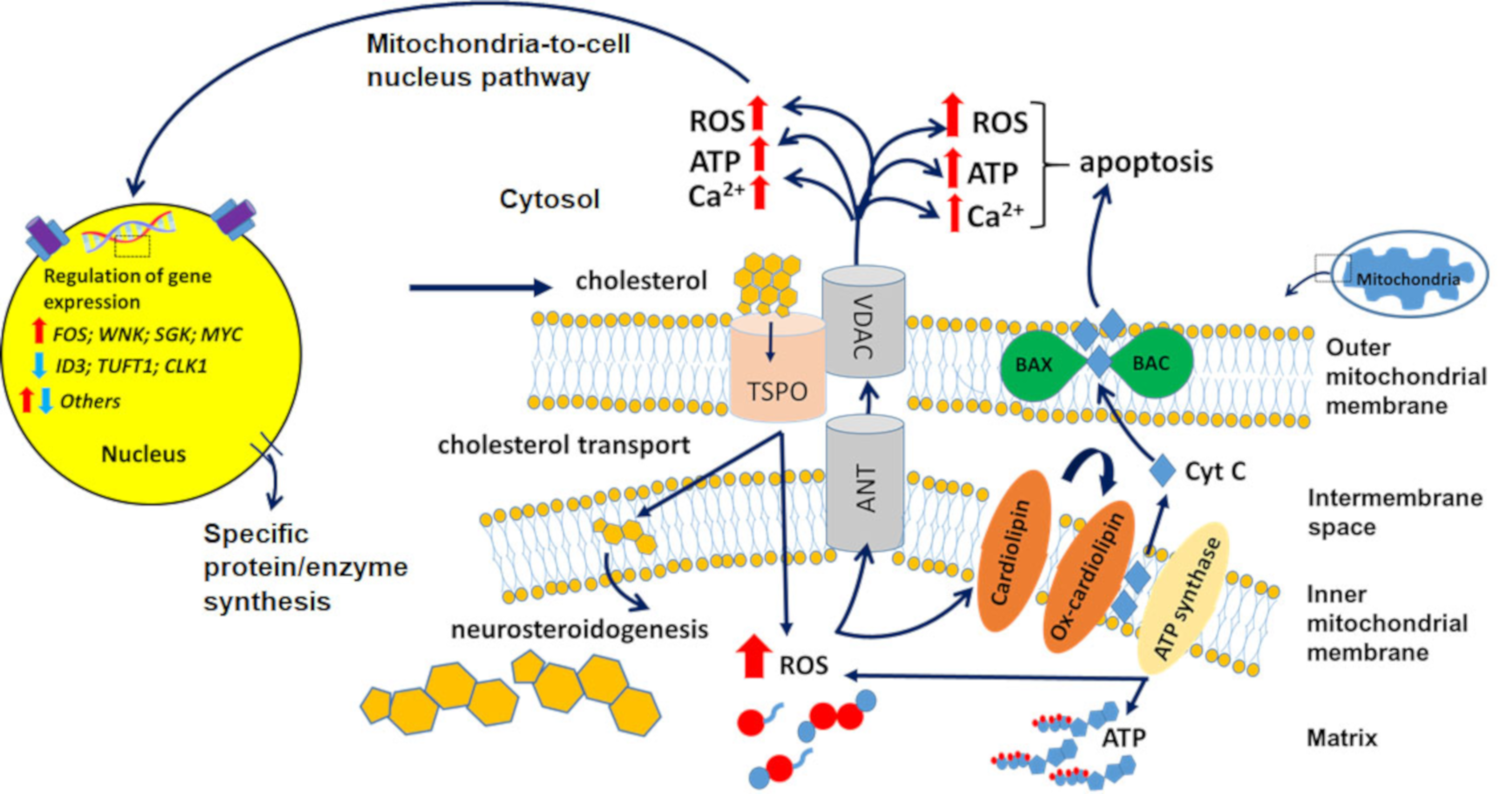
3. 18-kDa Translocator Protein (TSPO) as a Venue for Diagnostic and Therapeutic Tools for Brain Disorders
3.1. Neurons and TSPO
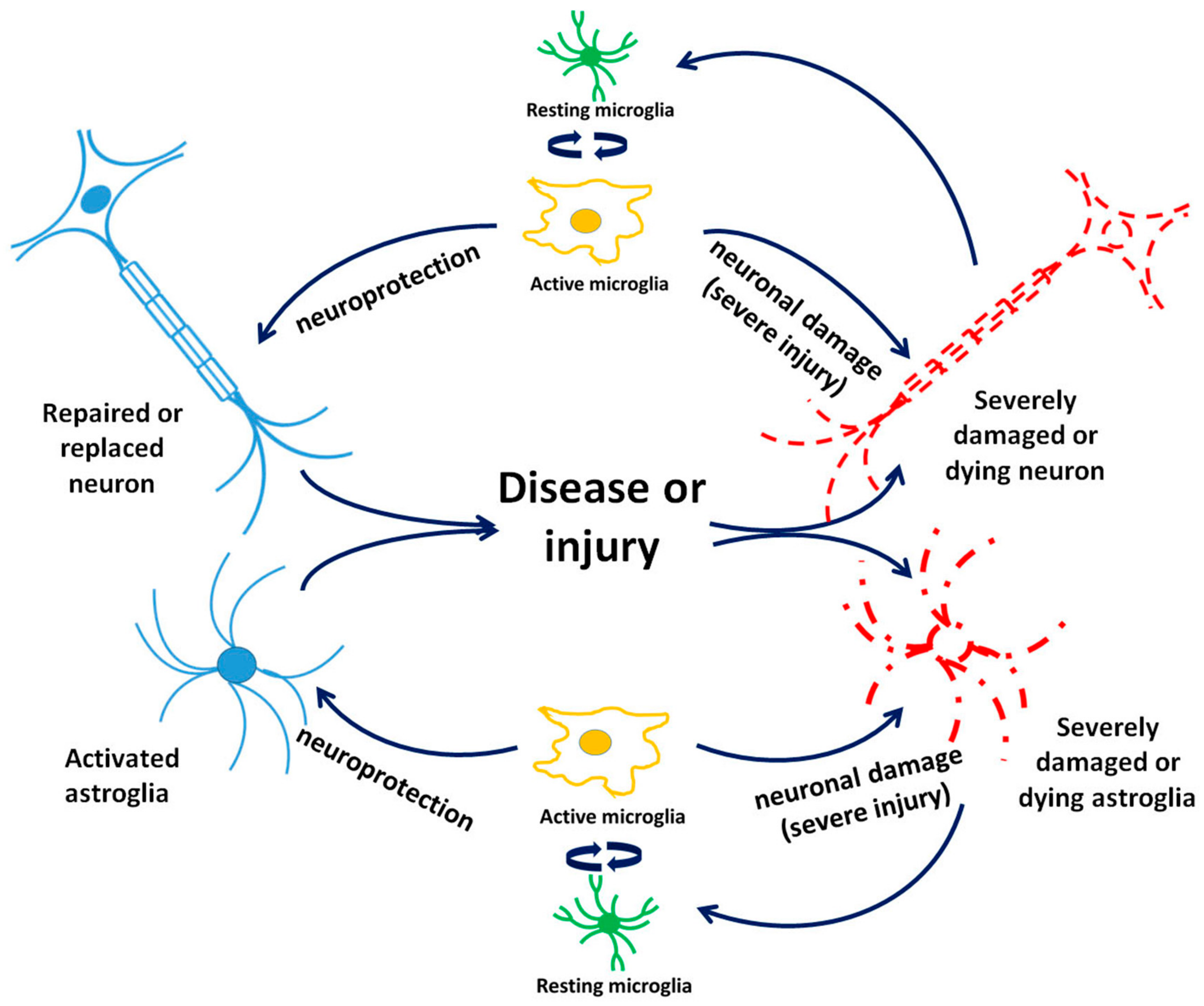
3.2. Astrocytes and TSPO
3.3. Microglia and TSPO
4. TSPO in Neurodegenerative Diseases
4.1. TSPO and Parkinson Disease (PD)
4.2. TSPO and Alzheimer’s Disease (AD)
4.3. TSPO and Huntington Disease (HD)
4.4. TSPO in Psychiatric Disorders
4.4.1. TSPO in Schizophrenia
4.4.2. TSPO in Autism Spectrum Disorder (ASD)
4.5. TSPO, Neurodegeneration, and Psychiatric Disorders in Short
5. TSPO and Alcohol Use Disorder (AUD)
AUD Excerpt: Mitochondrial Involvement, Including TSPO
6. TSPO and Traumatic Brain Injuries (TBIs)
7. TSPO and Stroke
8. Conclusions and Implications for Future Studies
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Gavish, M.; Bachman, I.; Shoukrun, R.; Katz, Y.; Veenman, L.; Weisinger, G.; Weizman, A. Enigma of the peripheral benzodiazepine receptor. Pharmacol. Rev. 1999, 51, 629–650. [Google Scholar] [PubMed]
- Papadopoulos, V.; Lecanu, L.; Brown, R.C.; Han, Z.; Yao, Z.X. Peripheral-type benzodiazepine receptor in neurosteroid biosynthesis, neuropathology and neurological disorders. Neuroscience 2006, 138, 749–756. [Google Scholar] [CrossRef] [PubMed]
- Gavish, M.; Weizman, R. Role of peripheral-type benzodiazepine receptors in steroidogenesis. Clin. Neuropharmacol. 1997, 20, 473–481. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Dimitrova-Shumkovska, J.; Veenman, L.; Ristoski, T.; Leschiner, S.; Gavish, M. Dimethylbenz[alpha]anthracene induces oxidative stress and reduces the binding capacity of the mitochondrial 18-kDa translocator protein in rat aorta. Drug Chem. Toxicol. 2010, 33, 337–347. [Google Scholar] [CrossRef]
- Dimitrova-Shumkovska, J.; Veenman, L.; Roim, I.; Leschiner, S.; Gavish, M. Thrombosis, Atherosclerosis and Atherothrombosis—New Insights and Experimental Protocols. In Thrombosis, Atherosclerosis and Atherothrombosis—New Insights and Experimental Protocols; Bozic-Mijovski, M., Ed.; InTech: Rijeka, Croatia, 2015. [Google Scholar]
- Veenman, L.; Alten, J.; Linnemannstons, K.; Shandalov, Y.; Zeno, S.; Lakomek, M.; Gavish, M.; Kugler, W. Potential involvement of F0F1-ATP(synth)ase and reactive oxygen species in apoptosis induction by the antineoplastic agent erucylphosphohomocholine in glioblastoma cell lines: A mechanism for induction of apoptosis via the 18 kDa mitochondrial translocator protein. Apoptosis 2010, 15, 753–768. [Google Scholar] [CrossRef] [Green Version]
- Veenman, L.; Bode, J.; Gaitner, M.; Caballero, B.; Pe’er, Y.; Zeno, S.; Kietz, S.; Kugler, W.; Lakomek, M.; Gavish, M. Effects of 18-kDa translocator protein knockdown on gene expression of glutamate receptors, transporters, and metabolism, and on cell viability affected by glutamate. Pharmacogenet. Genomics 2012, 22, 606–619. [Google Scholar] [CrossRef]
- Yasin, N.; Veenman, L.; Dimitrova-Shumkovska, J.; Gavish, M. The 18 kDa translocator protein, non-coding RNA, and homeostasis. Non-coding RNA Investigation 2017, 1, 25. [Google Scholar] [CrossRef]
- Zeno, S.; Veenman, L.; Katz, Y.; Bode, J.; Gavish, M.; Zaaroor, M. The 18 kDa mitochondrial translocator protein (TSPO) prevents accumulation of protoporphyrin IX. Involvement of reactive oxygen species (ROS). Curr. Mol. Med. 2012, 12, 494–501. [Google Scholar]
- Zeno, S.; Zaaroor, M.; Leschiner, S.; Veenman, L.; Gavish, M. CoCl(2) induces apoptosis via the 18 kDa translocator protein in U118MG human glioblastoma cells. Biochemistry 2009, 48, 4652–4661. [Google Scholar] [CrossRef]
- Veenman, L.; Shandalov, Y.; Gavish, M. VDAC activation by the 18 kDa translocator protein (TSPO), implications for apoptosis. J. Bioenerg. Biomembr. 2008, 40, 199–205. [Google Scholar] [CrossRef] [PubMed]
- Yasin, N.; Veenman, L.; Singh, S.; Azrad, M.; Bode, J.; Vainshtein, A.; Caballero, B.; Marek, I.; Gavish, M. Classical and Novel TSPO Ligands for the Mitochondrial TSPO Can Modulate Nuclear Gene Expression: Implications for Mitochondrial Retrograde Signaling. Int. J. Mol. Sci. 2017, 18, 786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caballero, B.; Veenman, L.; Gavish, M. Role of mitochondrial translocator protein (18 kDa) on mitochondrial-related cell death processes. Recent Pat. Endocr. Metab. Immune Drug Discov. 2013, 7, 86–101. [Google Scholar] [CrossRef] [PubMed]
- Kugler, W.; Veenman, L.; Shandalov, Y.; Leschiner, S.; Spanier, I.; Lakomek, M.; Gavish, M. Ligands of the mitochondrial 18 kDa translocator protein attenuate apoptosis of human glioblastoma cells exposed to erucylphosphohomocholine. Cell. Oncol. 2008, 30, 435–450. [Google Scholar] [PubMed]
- Veenman, L.; Gavish, M. Peripheral-type benzodiazepine receptors: Their implication in brain disease. Drug Dev. Res. 2000, 50, 355–370. [Google Scholar] [CrossRef]
- Khalil, B.; El Fissi, N.; Aouane, A.; Cabirol-Pol, M.J.; Rival, T.; Liévens, J.C. PINK1-induced mitophagy promotes neuroprotection in Huntington’s disease. Cell Death Dis. 2015, 6, e1617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, X.; Sun, X.; Starovoytov, V.; Cai, Q. Parkin-mediated mitophagy in mutant hAPP neurons and Alzheimer’s disease patient brains. Hum. Mol. Genet. 2015, 24, 2938–2951. [Google Scholar] [CrossRef]
- Veiga, S.; Azcoitia, I.; Garcia-Segura, L.M. Ro5-4864, a peripheral benzodiazepine receptor ligand, reduces reactive gliosis and protects hippocampal hilar neurons from kainic acid excitotoxicity. J. Neurosci. Res. 2005, 80, 129–137. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.K.; Guilarte, T.R. Translocator protein 18 kDa (TSPO): Molecular sensor of brain injury and repair. Pharmacol. Ther. 2008, 118, 1–17. [Google Scholar] [CrossRef]
- Chauveau, F.; Boutin, H.; Van Camp, N.; Dolle, F.; Tavitian, B. Nuclear imaging of neuroinflammation: A comprehensive review of [11C]PK11195 challengers. Eur. J. Nucl. Med. Mol. Imaging 2008, 35, 2304–2319. [Google Scholar] [CrossRef]
- Notter, T.; Coughlin, J.M.; Sawa, A.; Meyer, U. Reconceptualization of translocator protein as a biomarker of neuroinflammation in psychiatry. Mol. Psychiatry 2018, 23, 36–47. [Google Scholar] [CrossRef] [PubMed]
- Karchewski, L.A.; Bloechlinger, S.; Woolf, C.J. Axonal injury-dependent induction of the peripheral benzodiazepine receptor in small-diameter adult rat primary sensory neurons. Eur. J. Neurosci. 2004, 20, 671–683. [Google Scholar] [CrossRef] [PubMed]
- Mills, C.D.; Bitler, J.L.; Woolf, C.J. Role of the peripheral benzodiazepine receptor in sensory neuron regeneration. Mol. Cell. Neurosci. 2005, 30, 228–237. [Google Scholar] [CrossRef] [PubMed]
- Varga, B.; Marko, K.; Hadinger, N.; Jelitai, M.; Demeter, K.; Tihanyi, K.; Vas, A.; Madarasz, E. Translocator protein (TSPO 18kDa) is expressed by neural stem and neuronal precursor cells. Neurosci. Lett. 2009, 462, 257–262. [Google Scholar] [CrossRef] [PubMed]
- Mages, K.; Grassmann, F.; Jagle, H.; Rupprecht, R.; Weber, B.H.F.; Hauck, S.M.; Grosche, A. The agonistic TSPO ligand XBD173 attenuates the glial response thereby protecting inner retinal neurons in a murine model of retinal ischemia. J. Neuroinflamm. 2019, 16, 43. [Google Scholar] [CrossRef] [Green Version]
- Werry, E.L.; Bright, F.M.; Piguet, O.; Ittner, L.M.; Halliday, G.M.; Hodges, J.R.; Kiernan, M.C.; Loy, C.T.; Kril, J.J.; Kassiou, M. Recent Developments in TSPO PET Imaging as A Biomarker of Neuroinflammation in Neurodegenerative Disorders. Int. J. Mol. Sci. 2019, 20, E3161. [Google Scholar] [CrossRef] [Green Version]
- Vainshtein, A.; Veenman, L.; Shterenberg, A.; Singh, S.; Masarwa, A.; Dutta, B.; Island, B.; Tsoglin, E.; Levin, E.; Leschiner, S.; et al. Quinazoline-based tricyclic compounds that regulate programmed cell death, induce neuronal differentiation, and are curative in animal models for excitotoxicity and hereditary brain disease. Cell Death Discov. 2015, 1, 15027. [Google Scholar] [CrossRef]
- Liddelow, S.A.; Barres, B.A. Reactive Astrocytes: Production, Function, and Therapeutic Potential. Immunity 2017, 46, 957–967. [Google Scholar] [CrossRef] [Green Version]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Münch, A.E.; Chung, W.-S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef]
- Ren, Z.; Iliff, J.J.; Yang, L.; Yang, J.; Chen, X.; Chen, M.J.; Giese, R.N.; Wang, B.; Shi, X.; Nedergaard, M. ‘Hit & Run’ model of closed-skull traumatic brain injury (TBI) reveals complex patterns of post-traumatic AQP4 dysregulation. J. Cereb. Blood Flow Metab. 2013, 33, 834–845. [Google Scholar] [CrossRef] [PubMed]
- Zamanian, J.L.; Xu, L.; Foo, L.C.; Nouri, N.; Zhou, L.; Giffard, R.G.; Barres, B.A. Genomic analysis of reactive astrogliosis. J. Neurosci. 2012, 32, 6391–6410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hazell, A.S.; Desjardins, P.; Butterworth, R.F. Chronic exposure of rat primary astrocyte cultures to manganese results in increased binding sites for the ‘peripheral-type’ benzodiazepine receptor ligand 3H-PK 11195. Neurosci. Lett. 1999, 271, 5–8. [Google Scholar] [CrossRef]
- Lavisse, S.; Guillermier, M.; Herard, A.S.; Petit, F.; Delahaye, M.; Van Camp, N.; Ben Haim, L.; Lebon, V.; Remy, P.; Dolle, F.; et al. Reactive astrocytes overexpress TSPO and are detected by TSPO positron emission tomography imaging. J. Neurosci. 2012, 32, 10809–10818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carter, S.F.; Scholl, M.; Almkvist, O.; Wall, A.; Engler, H.; Langstrom, B.; Nordberg, A. Evidence for astrocytosis in prodromal Alzheimer disease provided by 11C-deuterium-L-deprenyl: a multitracer PET paradigm combining 11C-Pittsburgh compound B and 18F-FDG. J. Nucl. Med. 2012, 53, 37–46. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez-Vieitez, E.; Saint-Aubert, L.; Carter, S.F.; Almkvist, O.; Farid, K.; Scholl, M.; Chiotis, K.; Thordardottir, S.; Graff, C.; Wall, A.; et al. Diverging longitudinal changes in astrocytosis and amyloid PET in autosomal dominant Alzheimer’s disease. Brain 2016, 139, 922–936. [Google Scholar] [CrossRef] [Green Version]
- Venneti, S.; Lopresti, B.J.; Wiley, C.A. Molecular imaging of microglia/macrophages in the brain. Glia 2013, 61, 10–23. [Google Scholar] [CrossRef] [Green Version]
- Edison, P.; Donat, C.K.; Sastre, M. In vivo Imaging of Glial Activation in Alzheimer’s Disease. Front. Neurol. 2018, 9, 625. [Google Scholar] [CrossRef] [Green Version]
- Pannell, M.; Economopoulos, V.; Wilson, T.C.; Kersemans, V.; Isenegger, P.G.; Larkin, J.R.; Smart, S.; Gilchrist, S.; Gouverneur, V.; Sibson, N.R. Imaging of translocator protein upregulation is selective for pro-inflammatory polarized astrocytes and microglia. Glia 2020, 68, 280–297. [Google Scholar] [CrossRef] [Green Version]
- Del Rio-Hortega, P. Studies on neuroglia: Glia with very few processes (oligodendroglia) by PA-o del RA-o-Hortega. 1921. Clin. Neuropathol. 2012, 31, 440–459. [Google Scholar]
- Tronel, C.; Largeau, B.; Santiago Ribeiro, M.J.; Guilloteau, D.; Dupont, A.C.; Arlicot, N. Molecular Targets for PET Imaging of Activated Microglia: The Current Situation and Future Expectations. Int. J. Mol. Sci. 2017, 18, E802. [Google Scholar] [CrossRef] [Green Version]
- Colonna, M.; Butovsky, O. Microglia Function in the Central Nervous System During Health and Neurodegeneration. Annu. Rev. Immunol. 2017, 35, 441–468. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, C.L.; Martinez-Cerdeno, V.; Noctor, S.C. Microglia regulate the number of neural precursor cells in the developing cerebral cortex. J. Neurosci. 2013, 33, 4216–4233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paolicelli, R.C.; Bolasco, G.; Pagani, F.; Maggi, L.; Scianni, M.; Panzanelli, P.; Giustetto, M.; Ferreira, T.A.; Guiducci, E.; Dumas, L.; et al. Synaptic pruning by microglia is necessary for normal brain development. Science 2011, 333, 1456–1458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Squarzoni, P.; Oller, G.; Hoeffel, G.; Pont-Lezica, L.; Rostaing, P.; Low, D.; Bessis, A.; Ginhoux, F.; Garel, S. Microglia modulate wiring of the embryonic forebrain. Cell Rep. 2014, 8, 1271–1279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonsack, F.t.; Alleyne, C.H., Jr.; Sukumari-Ramesh, S. Augmented expression of TSPO after intracerebral hemorrhage: A role in inflammation? J. Neuroinflamm. 2016, 13, 151. [Google Scholar] [CrossRef] [Green Version]
- Monga, S.; Nagler, R.; Amara, R.; Weizman, A.; Gavish, M. Inhibitory Effects of the Two Novel TSPO Ligands 2-Cl-MGV-1 and MGV-1 on LPS-induced Microglial Activation. Cells 2019, 8, E486. [Google Scholar] [CrossRef] [Green Version]
- Owen, D.R.; Narayan, N.; Wells, L.; Healy, L.; Smyth, E.; Rabiner, E.A.; Galloway, D.; Williams, J.B.; Lehr, J.; Mandhair, H.; et al. Pro-inflammatory activation of primary microglia and macrophages increases 18 kDa translocator protein expression in rodents but not humans. J. Cereb. Blood Flow Metab. 2017, 37, 2679–2690. [Google Scholar] [CrossRef] [Green Version]
- Best, L.; Ghadery, C.; Pavese, N.; Tai, Y.F.; Strafella, A.P. New and Old TSPO PET Radioligands for Imaging Brain Microglial Activation in Neurodegenerative Disease. Curr. Neurol. Neurosci. Rep. 2019, 19, 24. [Google Scholar] [CrossRef]
- Donat, C.K.; Scott, G.; Gentleman, S.M.; Sastre, M. Microglial Activation in Traumatic Brain Injury. Front. Aging Neurosci. 2017, 9, 208. [Google Scholar] [CrossRef] [Green Version]
- Guilarte, T.R. TSPO in diverse CNS pathologies and psychiatric disease: A critical review and a way forward. Pharmacol. Ther. 2019, 194, 44–58. [Google Scholar] [CrossRef]
- Orihuela, R.; McPherson, C.A.; Harry, G.J. Microglial M1/M2 polarization and metabolic states. Br. J. Pharmacol. 2016, 173, 649–665. [Google Scholar] [CrossRef] [PubMed]
- Jellinger, K.A. Neuropathology of sporadic Parkinson’s disease: Evaluation and changes of concepts. Mov. Disord. 2012, 27, 8–30. [Google Scholar] [CrossRef] [PubMed]
- Maiti, P.; Manna, J.; Dunbar, G.L. Current understanding of the molecular mechanisms in Parkinson’s disease: Targets for potential treatments. Transl. Neurodegener. 2017, 6, 28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kempuraj, D.; Thangavel, R.; Selvakumar, G.P.; Zaheer, S.; Ahmed, M.E.; Raikwar, S.P.; Zahoor, H.; Saeed, D.; Natteru, P.A.; Iyer, S.; et al. Brain and Peripheral Atypical Inflammatory Mediators Potentiate Neuroinflammation and Neurodegeneration. Front. Cell. Neurosci. 2017, 11, 216. [Google Scholar] [CrossRef] [PubMed]
- Thome, A.D.; Harms, A.S.; Volpicelli-Daley, L.A.; Standaert, D.G. microRNA-155 Regulates Alpha-Synuclein-Induced Inflammatory Responses in Models of Parkinson Disease. J. Neurosci. 2016, 36, 2383–2390. [Google Scholar] [CrossRef]
- Yao, L.; Ye, Y.; Mao, H.; Lu, F.; He, X.; Lu, G.; Zhang, S. MicroRNA-124 regulates the expression of MEKK3 in the inflammatory pathogenesis of Parkinson’s disease. J. Neuroinflamm. 2018, 15, 13. [Google Scholar] [CrossRef] [Green Version]
- Gerhard, A.; Pavese, N.; Hotton, G.; Turkheimer, F.; Es, M.; Hammers, A.; Eggert, K.; Oertel, W.; Banati, R.B.; Brooks, D.J. In vivo imaging of microglial activation with [11C](R)-PK11195 PET in idiopathic Parkinson’s disease. Neurobiol. Dis. 2006, 21, 404–412. [Google Scholar] [CrossRef]
- Iannaccone, S.; Cerami, C.; Alessio, M.; Garibotto, V.; Panzacchi, A.; Olivieri, S.; Gelsomino, G.; Moresco, R.M.; Perani, D. In vivo microglia activation in very early dementia with Lewy bodies, comparison with Parkinson’s disease. Parkinsonism Relat. Disord. 2013, 19, 47–52. [Google Scholar] [CrossRef] [Green Version]
- Ouchi, Y.; Yoshikawa, E.; Sekine, Y.; Futatsubashi, M.; Kanno, T.; Ogusu, T.; Torizuka, T. Microglial activation and dopamine terminal loss in early Parkinson’s disease. Ann. Neurol. 2005, 57, 168–175. [Google Scholar] [CrossRef]
- Stokholm, M.G.; Iranzo, A.; Ostergaard, K.; Serradell, M.; Otto, M.; Svendsen, K.B.; Garrido, A.; Vilas, D.; Borghammer, P.; Santamaria, J.; et al. Assessment of neuroinflammation in patients with idiopathic rapid-eye-movement sleep behaviour disorder: A case-control study. Lancet Neurol. 2017, 16, 789–796. [Google Scholar] [CrossRef] [Green Version]
- Ghadery, C.; Koshimori, Y.; Coakeley, S.; Harris, M.; Rusjan, P.; Kim, J.; Houle, S.; Strafella, A.P. Microglial activation in Parkinson’s disease using [(18)F]-FEPPA. J. Neuroinflamm. 2017, 14, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varnas, K.; Cselenyi, Z.; Jucaite, A.; Halldin, C.; Svenningsson, P.; Farde, L.; Varrone, A. PET imaging of [(11)C]PBR28 in Parkinson’s disease patients does not indicate increased binding to TSPO despite reduced dopamine transporter binding. Eur. J. Nucl. Med. Mol. Imaging 2019, 46, 367–375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rizzo, G.; Veronese, M.; Tonietto, M.; Zanotti-Fregonara, P.; Turkheimer, F.E.; Bertoldo, A. Kinetic modeling without accounting for the vascular component impairs the quantification of [(11)C]PBR28 brain PET data. J. Cereb. Blood Flow Metab. 2014, 34, 1060–1069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, J.; Szego, E.M.; Leonov, A.; Benito, E.; Becker, S.; Fischer, A.; Zweckstetter, M.; Outeiro, T.; Schneider, A. Translocator Protein Ligand Protects against Neurodegeneration in the MPTP Mouse Model of Parkinsonism. J. Neurosci. 2019, 39, 3752–3769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azrad, M.; Zeineh, N.; Weizman, A.; Veenman, L.; Gavish, M. The TSPO Ligands 2-Cl-MGV-1, MGV-1, and PK11195 Differentially Suppress the Inflammatory Response of BV-2 Microglial Cell to LPS. Int. J. Mol. Sci. 2019, 20, 594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klein, W.L.; Krafft, G.A.; Finch, C.E. Targeting small Abeta oligomers: The solution to an Alzheimer’s disease conundrum? Trends Neurosci. 2001, 24, 219–224. [Google Scholar] [CrossRef]
- Qiu, K.; Zhang, X.; Wang, S.; Li, C.; Wang, X.; Li, X.; Wu, Y. TMP21 in Alzheimer’s Disease: Molecular Mechanisms and a Potential Target. Front. Cell. Neurosci. 2019, 13, 328. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Wu, Y.; Cai, F.; Song, W. Regulation of global gene expression in brain by TMP21. Mol. Brain 2019, 12, 39. [Google Scholar] [CrossRef]
- Xu, X.; Gao, H.; Qin, J.; He, L.; Liu, W. TMP21 modulates cell growth in papillary thyroid cancer cells by inducing autophagy through activation of the AMPK/mTOR pathway. Int. J. Clin. Exp. Pathol. 2015, 8, 10824–10831. [Google Scholar]
- Heneka, M.T.; Carson, M.J.; El Khoury, J.; Landreth, G.E.; Brosseron, F.; Feinstein, D.L.; Jacobs, A.H.; Wyss-Coray, T.; Vitorica, J.; Ransohoff, R.M.; et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015, 14, 388–405. [Google Scholar] [CrossRef] [Green Version]
- Cagnin, A.; Brooks, D.J.; Kennedy, A.M.; Gunn, R.N.; Myers, R.; Turkheimer, F.E.; Jones, T.; Banati, R.B. In-vivo measurement of activated microglia in dementia. Lancet 2001, 358, 461–467. [Google Scholar] [CrossRef]
- Venneti, S.; Lopresti, B.J.; Wang, G.; Hamilton, R.L.; Mathis, C.A.; Klunk, W.E.; Apte, U.M.; Wiley, C.A. PK11195 labels activated microglia in Alzheimer’s disease and in vivo in a mouse model using PET. Neurobiol. Aging 2009, 30, 1217–1226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Golla, S.S.; Boellaard, R.; Oikonen, V.; Hoffmann, A.; van Berckel, B.N.; Windhorst, A.D.; Virta, J.; Haaparanta-Solin, M.; Luoto, P.; Savisto, N.; et al. Quantification of [18F]DPA-714 binding in the human brain: initial studies in healthy controls and Alzheimer’s disease patients. J. Cereb. Blood Flow Metab. 2015, 35, 766–772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schuitemaker, A.; van der Doef, T.F.; Boellaard, R.; van der Flier, W.M.; Yaqub, M.; Windhorst, A.D.; Barkhof, F.; Jonker, C.; Kloet, R.W.; Lammertsma, A.A.; et al. Microglial activation in healthy aging. Neurobiol. Aging 2012, 33, 1067–1072. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Le, K.X.; Park, M.A.; Wang, S.; Belanger, A.P.; Dubey, S.; Frost, J.L.; Holton, P.; Reiser, V.; Jones, P.A.; et al. In Vivo Detection of Age- and Disease-Related Increases in Neuroinflammation by 18F-GE180 TSPO MicroPET Imaging in Wild-Type and Alzheimer’s Transgenic Mice. J. Neurosci. 2015, 35, 15716–15730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lejri, I.; Grimm, A.; Halle, F.; Abarghaz, M.; Klein, C.; Maitre, M.; Schmitt, M.; Bourguignon, J.J.; Mensah-Nyagan, A.G.; Bihel, F.; et al. TSPO Ligands Boost Mitochondrial Function and Pregnenolone Synthesis. J. Alzheimers Dis. 2019, 72, 1045–1058. [Google Scholar] [CrossRef] [Green Version]
- Vonsattel, J.P.; DiFiglia, M. Huntington disease. J. Neuropathol. Exp. Neurol. 1998, 57, 369–384. [Google Scholar] [CrossRef] [Green Version]
- Crotti, A.; Benner, C.; Kerman, B.E.; Gosselin, D.; Lagier-Tourenne, C.; Zuccato, C.; Cattaneo, E.; Gage, F.H.; Cleveland, D.W.; Glass, C.K. Mutant Huntingtin promotes autonomous microglia activation via myeloid lineage-determining factors. Nat. Neurosci. 2014, 17, 513–521. [Google Scholar] [CrossRef] [Green Version]
- Labbadia, J.; Morimoto, R.I. Huntington’s disease: Underlying molecular mechanisms and emerging concepts. Trends Biochem. Sci. 2013, 38, 378–385. [Google Scholar] [CrossRef] [Green Version]
- Pavese, N.; Gerhard, A.; Tai, Y.F.; Ho, A.K.; Turkheimer, F.; Barker, R.A.; Brooks, D.J.; Piccini, P. Microglial activation correlates with severity in Huntington disease: A clinical and PET study. Neurology 2006, 66, 1638–1643. [Google Scholar] [CrossRef]
- Politis, M.; Lahiri, N.; Niccolini, F.; Su, P.; Wu, K.; Giannetti, P.; Scahill, R.I.; Turkheimer, F.E.; Tabrizi, S.J.; Piccini, P. Increased central microglial activation associated with peripheral cytokine levels in premanifest Huntington’s disease gene carriers. Neurobiol. Dis. 2015, 83, 115–121. [Google Scholar] [CrossRef] [PubMed]
- Lois, C.; Gonzalez, I.; Izquierdo-Garcia, D.; Zurcher, N.R.; Wilkens, P.; Loggia, M.L.; Hooker, J.M.; Rosas, H.D. Neuroinflammation in Huntington’s Disease: New Insights with (11)C-PBR28 PET/MRI. ACS Chem. Neurosci. 2018, 9, 2563–2571. [Google Scholar] [CrossRef] [PubMed]
- Simmons, D.A.; James, M.L.; Belichenko, N.P.; Semaan, S.; Condon, C.; Kuan, J.; Shuhendler, A.J.; Miao, Z.; Chin, F.T.; Longo, F.M. TSPO-PET imaging using [18F]PBR06 is a potential translatable biomarker for treatment response in Huntington’s disease: Preclinical evidence with the p75NTR ligand LM11A-31. Hum. Mol. Genet. 2018, 27, 2893–2912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veenman, L.; Vainshtein, A.; Gavish, M. TSPO as a target for treatments of diseases, including neuropathological disorders. Cell Death Dis. 2015, 6, e1911. [Google Scholar] [CrossRef] [Green Version]
- Van Os, J.; Kapur, S. Schizophrenia. Lancet 2009, 374, 635–645. [Google Scholar] [CrossRef]
- Millar, J.K.; Christie, S.; Anderson, S.; Lawson, D.; Hsiao-Wei Loh, D.; Devon, R.S.; Arveiler, B.; Muir, W.J.; Blackwood, D.H.; Porteous, D.J. Genomic structure and localisation within a linkage hotspot of Disrupted In Schizophrenia 1, a gene disrupted by a translocation segregating with schizophrenia. Mol. Psychiatry 2001, 6, 173–178. [Google Scholar] [CrossRef] [Green Version]
- Shao, L.; Lu, B.; Wen, Z.; Teng, S.; Wang, L.; Zhao, Y.; Wang, L.; Ishizuka, K.; Xu, X.; Sawa, A.; et al. Disrupted-in-Schizophrenia-1 (DISC1) protein disturbs neural function in multiple disease-risk pathways. Hum. Mol. Genet. 2017, 26, 2634–2648. [Google Scholar] [CrossRef] [Green Version]
- Hattori, T.; Shimizu, S.; Koyama, Y.; Emoto, H.; Matsumoto, Y.; Kumamoto, N.; Yamada, K.; Takamura, H.; Matsuzaki, S.; Katayama, T.; et al. DISC1 (disrupted-in-schizophrenia-1) regulates differentiation of oligodendrocytes. PLoS ONE 2014, 9, e88506. [Google Scholar] [CrossRef] [Green Version]
- Kubota, K.; Kumamoto, N.; Matsuzaki, S.; Hashimoto, R.; Hattori, T.; Okuda, H.; Takamura, H.; Takeda, M.; Katayama, T.; Tohyama, M. Dysbindin engages in c-Jun N-terminal kinase activity and cytoskeletal organization. Biochem. Biophys. Res. Commun. 2009, 379, 191–195. [Google Scholar] [CrossRef]
- Shimizu, S.; Koyama, Y.; Hattori, T.; Tachibana, T.; Yoshimi, T.; Emoto, H.; Matsumoto, Y.; Miyata, S.; Katayama, T.; Ito, A.; et al. DBZ, a CNS-specific DISC1 binding protein, positively regulates oligodendrocyte differentiation. Glia 2014, 62, 709–724. [Google Scholar] [CrossRef]
- Tohyama, M.; Miyata, S.; Hattori, T.; Shimizu, S.; Matsuzaki, S. Molecular basis of major psychiatric diseases such as schizophrenia and depression. Anat. Sci. Int. 2015, 90, 137–143. [Google Scholar] [CrossRef] [PubMed]
- Morgan, J.T.; Chana, G.; Pardo, C.A.; Achim, C.; Semendeferi, K.; Buckwalter, J.; Courchesne, E.; Everall, I.P. Microglial activation and increased microglial density observed in the dorsolateral prefrontal cortex in autism. Biol. Psychiatry 2010, 68, 368–376. [Google Scholar] [CrossRef] [PubMed]
- Radewicz, K.; Garey, L.J.; Gentleman, S.M.; Reynolds, R. Increase in HLA-DR immunoreactive microglia in frontal and temporal cortex of chronic schizophrenics. J. Neuropathol. Exp. Neurol. 2000, 59, 137–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, B.J.; Buckley, P.; Seabolt, W.; Mellor, A.; Kirkpatrick, B. Meta-analysis of cytokine alterations in schizophrenia: Clinical status and antipsychotic effects. Biol. Psychiatry 2011, 70, 663–671. [Google Scholar] [CrossRef]
- Bromander, S.; Anckarsäter, R.; Kristiansson, M.; Blennow, K.; Zetterberg, H.; Anckarsäter, H.; Wass, C.E. Changes in serum and cerebrospinal fluid cytokines in response to non-neurological surgery: An observational study. J. Neuroinflamm. 2012, 9, 242. [Google Scholar] [CrossRef] [Green Version]
- van Berckel, B.N.; Bossong, M.G.; Boellaard, R.; Kloet, R.; Schuitemaker, A.; Caspers, E.; Luurtsema, G.; Windhorst, A.D.; Cahn, W.; Lammertsma, A.A.; et al. Microglia activation in recent-onset schizophrenia: A quantitative (R)-[11C]PK11195 positron emission tomography study. Biol. Psychiatry 2008, 64, 820–822. [Google Scholar] [CrossRef]
- Kenk, M.; Selvanathan, T.; Rao, N.; Suridjan, I.; Rusjan, P.; Remington, G.; Meyer, J.H.; Wilson, A.A.; Houle, S.; Mizrahi, R. Imaging neuroinflammation in gray and white matter in schizophrenia: An in-vivo PET study with [18F]-FEPPA. Schizophr. Bull. 2015, 41, 85–93. [Google Scholar] [CrossRef] [Green Version]
- Takano, A.; Arakawa, R.; Ito, H.; Tateno, A.; Takahashi, H.; Matsumoto, R.; Okubo, Y.; Suhara, T. Peripheral benzodiazepine receptors in patients with chronic schizophrenia: A PET study with [11C]DAA1106. Int. J. Neuropsychopharmacol. 2010, 13, 943–950. [Google Scholar] [CrossRef] [Green Version]
- Van der Doef, T.F.; Doorduin, J.; van Berckel, B.N.M.; Cervenka, S. Assessing brain immune activation in psychiatric disorders: clinical and preclinical PET imaging studies of the 18-kDa translocator protein. Clin. Transl. Imaging 2015, 3, 449–460. [Google Scholar] [CrossRef] [Green Version]
- Doorduin, J.; de Vries, E.F.; Willemsen, A.T.; de Groot, J.C.; Dierckx, R.A.; Klein, H.C. Neuroinflammation in schizophrenia-related psychosis: A PET study. J. Nucl. Med. 2009, 50, 1801–1807. [Google Scholar] [CrossRef] [Green Version]
- Setiawan, E.; Wilson, A.A.; Mizrahi, R.; Rusjan, P.M.; Miler, L.; Rajkowska, G.; Suridjan, I.; Kennedy, J.L.; Rekkas, P.V.; Houle, S.; et al. Role of translocator protein density, a marker of neuroinflammation, in the brain during major depressive episodes. JAMA Psychiatry 2015, 72, 268–275. [Google Scholar] [CrossRef] [PubMed]
- Collste, K.; Plaven-Sigray, P.; Fatouros-Bergman, H.; Victorsson, P.; Schain, M.; Forsberg, A.; Amini, N.; Aeinehband, S.; Karolinska Schizophrenia Project (KaSP) Consortium; Erhardt, S.; et al. Lower levels of the glial cell marker TSPO in drug-naive first-episode psychosis patients as measured using PET and [(11)C]PBR28. Mol. Psychiatry 2017, 22, 850–856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pouget, J.G.; Goncalves, V.F.; Nurmi, E.L.; Laughlin, P.C.; Mallya, K.S.; McCracken, J.T.; Aman, M.G.; McDougle, C.J.; Scahill, L.; Misener, V.L.; et al. Investigation of TSPO variants in schizophrenia and antipsychotic treatment outcomes. Pharmacogenomics 2015, 16, 5–22. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, Y.; Su, Y.; You, Y.; Ma, Y.; Yang, G.; Song, Y.; Liu, X.; Wang, M.; Zhang, L.; et al. The metabolic side effects of 12 antipsychotic drugs used for the treatment of schizophrenia on glucose: A network meta-analysis. BMC Psychiatry 2017, 17, 373. [Google Scholar] [CrossRef] [Green Version]
- Gut, P.; Baeza-Raja, B.; Andersson, O.; Hasenkamp, L.; Hsiao, J.; Hesselson, D.; Akassoglou, K.; Verdin, E.; Hirschey, M.D.; Stainier, D.Y.R. Whole-organism screening for gluconeogenesis identifies activators of fasting metabolism. Nat. Chem. Biol. 2013, 9, 97–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.F.; Chien, Y.L.; Wu, C.T.; Shang, C.Y.; Wu, Y.Y.; Gau, S.S. Deficits in executive functions among youths with autism spectrum disorders: an age-stratified analysis. Psychol. Med. 2016, 46, 1625–1638. [Google Scholar] [CrossRef] [Green Version]
- Lai, M.-C.; Lombardo, M.V.; Baron-Cohen, S. Autism. Lancet 2014, 383, 896–910. [Google Scholar] [CrossRef]
- Kim, J.W.; Hong, J.Y.; Bae, S.M. Microglia and Autism Spectrum Disorder: Overview of Current Evidence and Novel Immunomodulatory Treatment Options. Clin. Psychopharmacol. Neurosci. 2018, 16, 246–252. [Google Scholar] [CrossRef]
- Takano, T. Role of Microglia in Autism: Recent Advances. Dev. Neurosci. 2015, 37, 195–202. [Google Scholar] [CrossRef]
- Tang, G.; Gudsnuk, K.; Kuo, S.H.; Cotrina, M.L.; Rosoklija, G.; Sosunov, A.; Sonders, M.S.; Kanter, E.; Castagna, C.; Yamamoto, A.; et al. Loss of mTOR-dependent macroautophagy causes autistic-like synaptic pruning deficits. Neuron 2014, 83, 1131–1143. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Ji, F.; Wang, G.; He, D.; Yang, L.; Zhang, M. BDNF Activates mTOR to Upregulate NR2B Expression in the Rostral Anterior Cingulate Cortex Required for Inflammatory Pain-Related Aversion in Rats. Neurochem. Res. 2018, 43, 681–691. [Google Scholar] [CrossRef] [PubMed]
- Edmonson, C.A.; Ziats, M.N.; Rennert, O.M. A Non-inflammatory Role for Microglia in Autism Spectrum Disorders. Front. Neurol. 2016, 7, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pardo, C.A.; Vargas, D.L.; Zimmerman, A.W. Immunity, neuroglia and neuroinflammation in autism. Int. Rev. Psychiatry 2005, 17, 485–495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, K.; Sugihara, G.; Ouchi, Y.; Nakamura, K.; Futatsubashi, M.; Takebayashi, K.; Yoshihara, Y.; Omata, K.; Matsumoto, K.; Tsuchiya, K.J.; et al. Microglial activation in young adults with autism spectrum disorder. JAMA Psychiatry 2013, 70, 49–58. [Google Scholar] [CrossRef] [PubMed]
- P.A.H.O (PAHO). Regional Status Report on Alcohol and Health in the Americas; Pan American Health Organization: Washington, DC, USA, 2015; ISBN 978-92-75-11855-9. [Google Scholar]
- LaVallee, R.A.; Williams, G.D.; Hsiao, Y.Y. Surveillance Report #87: Apparent per Capita Alcohol Consumption: National, State, and Regional trends, 1977–2007; National Institute on Alcohol Abuse and Alcoholism Arlington, VA, USA, 2007; National Institute on Alcohol Abuse and Alcoholism: Arlington, VA, USA, 2007. [Google Scholar]
- Rehm, J.; Mathers, C.; Popova, S.; Thavorncharoensap, M.; Teerawattananon, Y.; Patra, J. Global burden of disease and injury and economic cost attributable to alcohol use and alcohol-use disorders. Lancet 2009, 373, 2223. [Google Scholar] [CrossRef]
- Brust, J.C. Ethanol and cognition: Indirect effects, neurotoxicity and neuroprotection: A review. Int. J. Environ. Res. Public Health 2010, 7, 1540–1557. [Google Scholar] [CrossRef]
- Harper, C. The neurotoxicity of alcohol. Hum. Exp. Toxicol. 2007, 26, 251–257. [Google Scholar] [CrossRef]
- Mende, M.A. Alcohol in the Aging Brain—The Interplay Between Alcohol Consumption, Cognitive Decline and the Cardiovascular System. Front. Neurosci. 2019, 13, 713. [Google Scholar] [CrossRef]
- Ritchie, H.; Roser, M. Alcohol Consumption. Our World in Data 2020. “Alcohol Consumption”. 2020. Available online: https://ourworldindata.org/alcohol-consumption (accessed on 5 March 2020).
- Tapia-Rojas, C.; Mira, R.G.; Torres, A.K.; Jara, C.; Perez, M.J.; Vergara, E.H.; Cerpa, W.; Quintanilla, R.A. Alcohol consumption during adolescence: A link between mitochondrial damage and ethanol brain intoxication. Birth Defects Res. 2017, 109, 1623–1639. [Google Scholar] [CrossRef]
- Nakamura, K.; Iwahashi, K.; Furukawa, A.; Ameno, K.; Kinoshita, H.; Ijiri, I.; Sekine, Y.; Suzuki, K.; Iwata, Y.; Minabe, Y.; et al. Acetaldehyde adducts in the brain of alcoholics. Arch. Toxicol. 2003, 77, 591–593. [Google Scholar] [CrossRef]
- Upadhya, S.C.; Ravindranath, V. Detection and localization of protein-acetaldehyde adducts in rat brain after chronic ethanol treatment. Alcohol. Clin. Exp. Res. 2002, 26, 856–863. [Google Scholar] [CrossRef] [PubMed]
- Kokubo, J.; Nagatani, N.; Hiroki, K.; Kuroiwa, K.; Watanabe, N.; Arai, T. Mechanism of Destruction of Microtubule Structures by 4-Hydroxy-2-Nonenal. Cell Struct. Funct. 2008, 33, 51–59. [Google Scholar] [CrossRef] [Green Version]
- Stewart, B.J.; Doorn, J.A.; Petersen, D.R. Residue-specific adduction of tubulin by 4-hydroxynonenal and 4-oxononenal causes cross-linking and inhibits polymerization. Chem. Res. Toxicol. 2007, 20, 1111–1119. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Hu, P. Proanthocyanidins prevent ethanol-induced cognitive impairment by suppressing oxidative and inflammatory stress in adult rat brain. Neuroreport 2017, 28, 980–986. [Google Scholar] [CrossRef] [PubMed]
- Friedman, J.R.; Nunnari, J. Mitochondrial form and function. Nature 2014, 505, 335–343. [Google Scholar] [CrossRef] [Green Version]
- Sheng, Z.H. Mitochondrial trafficking and anchoring in neurons: New insight and implications. J. Cell Biol. 2014, 204, 1087–1098. [Google Scholar] [CrossRef] [Green Version]
- Grimm, A.; Eckert, A. Brain aging and neurodegeneration: from a mitochondrial point of view. J. Neurochem. 2017, 143, 418–431. [Google Scholar] [CrossRef] [Green Version]
- Estaquier, J.; Vallette, F.; Vayssiere, J.L.; Mignotte, B. The mitochondrial pathways of apoptosis. Adv. Exp. Med. Biol. 2012, 942, 157–183. [Google Scholar] [CrossRef]
- Giorgi, C.; Baldassari, F.; Bononi, A.; Bonora, M.; De Marchi, E.; Marchi, S.; Missiroli, S.; Patergnani, S.; Rimessi, A.; Suski, J.M.; et al. Mitochondrial Ca(2+) and apoptosis. Cell Calcium 2012, 52, 36–43. [Google Scholar] [CrossRef] [Green Version]
- Guadagnoli, T.; Caltana, L.; Vacotto, M.; Gironacci, M.; Brusco, A. Direct effects of ethanol on neuronal differentiation: An in vitro analysis of viability and morphology. Brain Res. Bull. 2016, 127, 177–186. [Google Scholar] [CrossRef]
- Perez, M.J.; Quintanilla, R.A. Development or disease: Duality of the mitochondrial permeability transition pore. Dev. Biol. 2017, 426, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Hunter, D.R.; Haworth, R.A. The Ca2+-induced membrane transition in mitochondria. I. The protective mechanisms. Arch. Biochem. Biophys. 1979, 195, 453–459. [Google Scholar] [CrossRef]
- Halestrap, A.P. What is the mitochondrial permeability transition pore? J. Mol. Cell. Cardiol. 2009, 46, 821–831. [Google Scholar] [CrossRef] [PubMed]
- Tsujimoto, Y.; Shimizu, S. Role of the mitochondrial membrane permeability transition in cell death. Apoptosis 2007, 12, 835–840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rao, V.K.; Carlson, E.A.; Yan, S.S. Mitochondrial permeability transition pore is a potential drug target for neurodegeneration. Biochim. Biophys. Acta 2014, 1842, 1267–1272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ozkol, H.; Bulut, G.; Balahoroglu, R.; Tuluce, Y.; Ozkol, H.U. Protective Effects of Selenium, N-Acetylcysteine and Vitamin E Against Acute Ethanol Intoxication in Rats. Biol. Trace Elem. Res. 2017, 175, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Lamarche, F.; Carcenac, C.; Gonthier, B.; Cottet-Rousselle, C.; Chauvin, C.; Barret, L.; Leverve, X.; Savasta, M.; Fontaine, E. Mitochondrial permeability transition pore inhibitors prevent ethanol-induced neuronal death in mice. Chem. Res. Toxicol. 2013, 26, 78–88. [Google Scholar] [CrossRef]
- Liu, Z.; Liu, Y.; Gao, R.; Li, H.; Dunn, T.; Wu, P.; Smith, R.G.; Sarkar, P.S.; Fang, X. Ethanol suppresses PGC-1α expression by interfering with the cAMP-CREB pathway in neuronal cells. PLoS ONE 2014, 9, e104247. [Google Scholar] [CrossRef]
- Lopez-Armada, M.J.; Riveiro-Naveira, R.R.; Vaamonde-Garcia, C.; Valcarcel-Ares, M.N. Mitochondrial dysfunction and the inflammatory response. Mitochondrion 2013, 13, 106–118. [Google Scholar] [CrossRef]
- Bode, J.; Veenman, L.; Vainshtein, A.; Kugler, W.; Rosenberg, B.J.; Gavish, M. Modulation of Gene Expression Associated with the Cell Cycle and Tumorigenicity of Glioblastoma Cells by the 18 kDa Translocator Protein (TSPO). Austin J. Pharmacol. Ther. 2014, 2, 1053. [Google Scholar]
- Gavish, M.; Veenman, L. Regulation of Mitochondrial, Cellular, and Organismal Functions by TSPO. Adv. Pharmacol. 2018, 82, 103–136. [Google Scholar] [CrossRef] [PubMed]
- Shargorodsky, L.; Veenman, L.; Caballero, B.; Pe’er, Y.; Leschiner, S.; Bode, J.; Gavish, M. The nitric oxide donor sodium nitroprusside requires the 18 kDa Translocator Protein to induce cell death. Apoptosis 2012, 17, 647–665. [Google Scholar] [CrossRef] [PubMed]
- Veenman, L.; Vainshtein, A.; Yasin, N.; Azrad, M.; Gavish, M. Tetrapyrroles as Endogenous TSPO Ligands in Eukaryotes and Prokaryotes: Comparisons with Synthetic Ligands. Int. J. Mol. Sci. 2016, 17, 880. [Google Scholar] [CrossRef] [PubMed]
- Fitzpatrick, L.E.; Jackson, M.; Crowe, S.F. The relationship between alcoholic cerebellar degeneration and cognitive and emotional functioning. Neurosci. Biobehav. Rev. 2008, 32, 466–485. [Google Scholar] [CrossRef] [PubMed]
- Marshall, E.J. Adolescent Alcohol Use: Risks and Consequences. Alcohol Alcohol. 2014, 49, 160–164. [Google Scholar] [CrossRef] [Green Version]
- Wackernah, R.C.; Minnick, M.J.; Clapp, P. Alcohol use disorder: pathophysiology, effects, and pharmacologic options for treatment. Subst. Abuse Rehabil. 2014, 5, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Alfonso-Loeches, S.; Urena-Peralta, J.; Morillo-Bargues, M.J.; Gomez-Pinedo, U.; Guerri, C. Ethanol-Induced TLR4/NLRP3 Neuroinflammatory Response in Microglial Cells Promotes Leukocyte Infiltration Across the BBB. Neurochem. Res. 2016, 41, 193–209. [Google Scholar] [CrossRef]
- Fernandez, G.M.; Lew, B.J.; Vedder, L.C.; Savage, L.M. Chronic intermittent ethanol exposure leads to alterations in brain-derived neurotrophic factor within the frontal cortex and impaired behavioral flexibility in both adolescent and adult rats. Neuroscience 2017, 348, 324–334. [Google Scholar] [CrossRef] [Green Version]
- Pla, A.; Pascual, M.; Guerri, C. Autophagy Constitutes a Protective Mechanism against Ethanol Toxicity in Mouse Astrocytes and Neurons. PLoS ONE 2016, 11, e0153097. [Google Scholar] [CrossRef] [Green Version]
- Maeda, J.; Higuchi, M.; Inaji, M.; Ji, B.; Haneda, E.; Okauchi, T.; Zhang, M.R.; Suzuki, K.; Suhara, T. Phase-dependent roles of reactive microglia and astrocytes in nervous system injury as delineated by imaging of peripheral benzodiazepine receptor. Brain Res. 2007, 1157, 100–111. [Google Scholar] [CrossRef]
- Ito, F.; Toyama, H.; Kudo, G.; Suzuki, H.; Hatano, K.; Ichise, M.; Katada, K.; Ito, K.; Sawada, M. Two activated stages of microglia and PET imaging of peripheral benzodiazepine receptors with [(11)C]PK11195 in rats. Ann. Nucl. Med. 2010, 24, 163–169. [Google Scholar] [CrossRef] [PubMed]
- Saba, W.; Goutal, S.; Auvity, S.; Kuhnast, B.; Coulon, C.; Kouyoumdjian, V.; Buvat, I.; Leroy, C.; Tournier, N. Imaging the neuroimmune response to alcohol exposure in adolescent baboons: A TSPO PET study using 18F-DPA-714. Addict. Biol. 2018, 23, 1000–1009. [Google Scholar] [CrossRef] [PubMed]
- Kalk, N.J.; Guo, Q.; Owen, D.; Cherian, R.; Erritzoe, D.; Gilmour, A.; Ribeiro, A.S.; McGonigle, J.; Waldman, A.; Matthews, P.; et al. Decreased hippocampal translocator protein (18 kDa) expression in alcohol dependence: a [(11)C]PBR28 PET study. Transl. Psychiatry 2017, 7, e996. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.W.; Wiers, C.E.; Tyler, R.; Shokri-Kojori, E. Influence of alcoholism and cholesterol on TSPO binding in brain: PET [(11)C]PBR28 studies in humans and rodents. Neuropsychopharmacology 2018, 43, 1832–1839. [Google Scholar] [CrossRef] [PubMed]
- Lin, R.; Rittenhouse, D.; Sweeney, K.; Potluri, P.; Wallace, D.C. TSPO, a Mitochondrial Outer Membrane Protein, Controls Ethanol-Related Behaviors in Drosophila. PLOS Genetics 2015, 11, e1005366. [Google Scholar] [CrossRef]
- Werner, C.; Engelhard, K. Pathophysiology of traumatic brain injury. Br. J. Anaesth. 2007, 99, 4–9. [Google Scholar] [CrossRef] [Green Version]
- Pastuhov, S.I.; Hisamoto, N.; Matsumoto, K. MAP kinase cascades regulating axon regeneration in C. elegans. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2015, 91, 63–75. [Google Scholar] [CrossRef] [Green Version]
- Jassam, Y.N.; Izzy, S.; Whalen, M.; McGavern, D.B.; El Khoury, J. Neuroimmunology of Traumatic Brain Injury: Time for a Paradigm Shift. Neuron 2017, 95, 1246–1265. [Google Scholar] [CrossRef] [Green Version]
- Shabab, T.; Khanabdali, R.; Moghadamtousi, S.Z.; Kadir, H.A.; Mohan, G. Neuroinflammation pathways: A general review. Int. J. Neurosci. 2017, 127, 624–633. [Google Scholar] [CrossRef]
- Finnie, J.W. Animal models of traumatic brain injury: A review. Aust. Vet. J. 2001, 79, 628–633. [Google Scholar] [CrossRef]
- Rosenberg, N.; Yasin, N.; Veenman, L.; Rosenberg, O.; Weizman, A.; Gavish, M. 18 kDa Translocator Protein in Mitochondria-Related Pathology: The Case of Traumatic Brain Injury. In Mitochondrial Diseases; Taskin, E., Ed.; IntechOpen: London, UK, 2018. [Google Scholar]
- Ma, L.; Zhang, H.; Liu, N.; Wang, P.Q.; Guo, W.Z.; Fu, Q.; Jiao, L.B.; Ma, Y.Q.; Mi, W.D. TSPO ligand PK11195 alleviates neuroinflammation and beta-amyloid generation induced by systemic LPS administration. Brain Res. Bull. 2016, 121, 192–200. [Google Scholar] [CrossRef] [PubMed]
- Scholz, R.; Caramoy, A.; Bhuckory, M.B.; Rashid, K.; Chen, M.; Xu, H.; Grimm, C.; Langmann, T. Targeting translocator protein (18 kDa) (TSPO) dampens pro-inflammatory microglia reactivity in the retina and protects from degeneration. J. Neuroinflamm. 2015, 12, 201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barron, A.M.; Garcia-Segura, L.M.; Caruso, D.; Jayaraman, A.; Lee, J.W.; Melcangi, R.C.; Pike, C.J. Ligand for translocator protein reverses pathology in a mouse model of Alzheimer’s disease. J. Neurosci. 2013, 33, 8891–8897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shehadeh, M.; Palzur, E.; Apel, L.; Soustiel, J.F. Reduction of Traumatic Brain Damage by Tspo Ligand Etifoxine. Int. J. Mol. Sci. 2019, 20, E2639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bader, S.; Wolf, L.; Milenkovic, V.M.; Gruber, M.; Nothdurfter, C.; Rupprecht, R.; Wetzel, C.H. Differential effects of TSPO ligands on mitochondrial function in mouse microglia cells. Psychoneuroendocrinology 2019, 106, 65–76. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.; Pae, A.N. Translocator protein (TSPO) ligands for the diagnosis or treatment of neurodegenerative diseases: a patent review (2010–2015; part 1). Expert Opin. Ther. Pat. 2016, 26, 1325–1351. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Veenman, L.; Singh, S.; Ouyang, F.; Liang, J.; Huang, W.; Marek, I.; Zeng, J.; Gavish, M. 2-Cl-MGV-1 Ameliorates Apoptosis in the Thalamus and Hippocampus and Cognitive Deficits After Cortical Infarct in Rats. Stroke 2017, 48, 3366–3374. [Google Scholar] [CrossRef]
- Zeineh, N.; Nagler, R.; Gabay, M.; Weizman, A.; Gavish, M. Effects of Cigarette Smoke on TSPO-related Mitochondrial Processes. Cells 2019, 8, E694. [Google Scholar] [CrossRef] [Green Version]
- Cao, T.; Thomas, T.C.; Ziebell, J.M.; Pauly, J.R.; Lifshitz, J. Morphological and genetic activation of microglia after diffuse traumatic brain injury in the rat. Neuroscience 2012, 225, 65–75. [Google Scholar] [CrossRef] [Green Version]
- Donat, C.K.; Gaber, K.; Meixensberger, J.; Brust, P.; Pinborg, L.H.; Hansen, H.H.; Mikkelsen, J.D. Changes in Binding of [(123)I]CLINDE, a High-Affinity Translocator Protein 18 kDa (TSPO) Selective Radioligand in a Rat Model of Traumatic Brain Injury. Neuromol. Med. 2016, 18, 158–169. [Google Scholar] [CrossRef] [Green Version]
- Grossman, R.; Paden, C.M.; Fry, P.A.; Rhodes, R.S.; Biegon, A. Persistent region-dependent neuroinflammation, NMDA receptor loss and atrophy in an animal model of penetrating brain injury. Future Neurol. 2012, 7, 329–339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Israel, I.; Ohsiek, A.; Al-Momani, E.; Albert-Weissenberger, C.; Stetter, C.; Mencl, S.; Buck, A.K.; Kleinschnitz, C.; Samnick, S.; Sirén, A.-L. Combined [(18)F]DPA-714 micro-positron emission tomography and autoradiography imaging of microglia activation after closed head injury in mice. J. Neuroinflamm. 2016, 13, 140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Yue, X.; Kiesewetter, D.O.; Niu, G.; Teng, G.; Chen, X. PET imaging of neuroinflammation in a rat traumatic brain injury model with radiolabeled TSPO ligand DPA-714. Eur. J. Nucl. Med. Mol. Imaging 2014, 41, 1440–1449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raghavendra Rao, V.L.; Dogan, A.; Bowen, K.K.; Dempsey, R.J. Traumatic brain injury leads to increased expression of peripheral-type benzodiazepine receptors, neuronal death, and activation of astrocytes and microglia in rat thalamus. Exp. Neurol. 2000, 161, 102–114. [Google Scholar] [CrossRef] [PubMed]
- Yu, I.; Inaji, M.; Maeda, J.; Okauchi, T.; Nariai, T.; Ohno, K.; Higuchi, M.; Suhara, T. Glial cell-mediated deterioration and repair of the nervous system after traumatic brain injury in a rat model as assessed by positron emission tomography. J. Neurotrauma 2010, 27, 1463–1475. [Google Scholar] [CrossRef]
- Kim, E.J.; Yu, S.W. Translocator protein 18 kDa (TSPO): Old dogma, new mice, new structure, and new questions for neuroprotection. Neural Regen. Res. 2015, 10, 878–880. [Google Scholar] [CrossRef]
- Schumacher, M.; Guennoun, R.; Stein, D.G.; De Nicola, A.F. Progesterone: therapeutic opportunities for neuroprotection and myelin repair. Pharmacol. Ther. 2007, 116, 77–106. [Google Scholar] [CrossRef]
- Price, C.; Wang, D.; Menon, D.; Guadagno, J.; Cleij, M.; Fryer, T.; Aigbirhio, F.; Baron, J.-C.; Warburton, E. Intrinsic activated microglia map to the peri-infarct zone in the subacute phase of ischemic stroke. Stroke 2006, 37, 1749–1753. [Google Scholar] [CrossRef] [Green Version]
- Gulyas, B.; Toth, M.; Schain, M.; Airaksinen, A.; Vas, A.; Kostulas, K.; Lindstrom, P.; Hillert, J.; Halldin, C. Evolution of microglial activation in ischaemic core and peri-infarct regions after stroke: a PET study with the TSPO molecular imaging biomarker [((11))C]vinpocetine. J. Neurol. Sci. 2012, 320, 110–117. [Google Scholar] [CrossRef]
- Martin, A.; Boisgard, R.; Kassiou, M.; Dolle, F.; Tavitian, B. Reduced PBR/TSPO expression after minocycline treatment in a rat model of focal cerebral ischemia: A PET study using [(18)F]DPA-714. Mol. Imaging Biol. 2011, 13, 10–15. [Google Scholar] [CrossRef]
- Boutin, H.; Murray, K.; Pradillo, J.; Maroy, R.; Smigova, A.; Gerhard, A.; Jones, P.A.; Trigg, W. 18F-GE-180: A novel TSPO radiotracer compared to 11C-R-PK11195 in a preclinical model of stroke. Eur. J. Nucl. Med. Mol. Imaging 2015, 42, 503–511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pulagam, K.R.; Colás, L.; Padro, D.; Plaza-García, S.; Gómez-Vallejo, V.; Higuchi, M.; Llop, J.; Martín, A. Evaluation of the novel TSPO radiotracer [(18)F] VUIIS1008 in a preclinical model of cerebral ischemia in rats. EJNMMI Res. 2017, 7, 93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaney, A.; Cropper, H.C.; Johnson, E.M.; Lechtenberg, K.J.; Peterson, T.C.; Stevens, M.Y.; Buckwalter, M.S.; James, M.L. (11)C-DPA-713 Versus (18)F-GE-180: A Preclinical Comparison of Translocator Protein 18 kDa PET Tracers to Visualize Acute and Chronic Neuroinflammation in a Mouse Model of Ischemic Stroke. J. Nucl. Med. 2019, 60, 122–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sridharan, S.; Lepelletier, F.X.; Trigg, W.; Banister, S.; Reekie, T.; Kassiou, M.; Gerhard, A.; Hinz, R.; Boutin, H. Comparative Evaluation of Three TSPO PET Radiotracers in a LPS-Induced Model of Mild Neuroinflammation in Rats. Mol. Imaging Biol. 2017, 19, 77–89. [Google Scholar] [CrossRef] [PubMed]
- Frostegård, J. Immunity, atherosclerosis and cardiovascular disease. BMC Med. 2013, 11, 117. [Google Scholar] [CrossRef] [Green Version]
- Foss, C.A.; Bedja, D.; Mease, R.C.; Wang, H.; Kass, D.A.; Chatterjee, S.; Pomper, M.G. Molecular imaging of inflammation in the ApoE-/- mouse model of atherosclerosis with IodoDPA. Biochem. Biophys. Res. Commun. 2015, 461, 70–75. [Google Scholar] [CrossRef] [Green Version]
- Grosse, G.M.; Bascunana, P.; Schulz-Schaeffer, W.J.; Teebken, O.E.; Wilhelmi, M.; Worthmann, H.; Ross, T.L.; Wester, H.J.; Kropf, S.; Derlin, T.; et al. Targeting Chemokine Receptor CXCR4 and Translocator Protein for Characterization of High-Risk Plaque in Carotid Stenosis Ex Vivo. Stroke 2018, 49, 1988–1991. [Google Scholar] [CrossRef]
- Chen, W.H.; Yeh, H.L.; Tsao, C.W.; Lien, L.M.; Chiwaya, A.; Alizargar, J.; Bai, C.H. Plasma Translocator Protein Levels and Outcomes of Acute Ischemic Stroke: A Pilot Study. Dis. Markers 2018, 2018, 9831079. [Google Scholar] [CrossRef]
- Li, M.; Ren, H.; Sheth, K.N.; Shi, F.D.; Liu, Q. A TSPO ligand attenuates brain injury after intracerebral hemorrhage. FASEB J. 2017, 31, 3278–3287. [Google Scholar] [CrossRef] [Green Version]
- Dimitrova-Shumkovska, J.; Krstanoski, L. Alpha-Synuclein Aggregation, Cholesterol Transport, and the 18-kDa Translocator Protein. In Synucleins-Biochemistry and Role in Diseases; Surguchov, A., Ed.; IntechOpen: London, UK, 2019. [Google Scholar]
- Li, H.D.; Li, M.; Shi, E.; Jin, W.N.; Wood, K.; Gonzales, R.; Liu, Q. A translocator protein 18 kDa agonist protects against cerebral ischemia/reperfusion injury. J. Neuroinflamm. 2017, 14, 151. [Google Scholar] [CrossRef] [Green Version]
- Henderson, F.; Hart, P.J.; Pradillo, J.M.; Kassiou, M.; Christie, L.; Williams, K.J.; Boutin, H.; McMahon, A. Multi-modal imaging of long-term recovery post-stroke by positron emission tomography and matrix-assisted laser desorption/ionisation mass spectrometry. Rapid Commun. Mass Spectrom. 2018, 32, 721–729. [Google Scholar] [CrossRef] [PubMed] [Green Version]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dimitrova-Shumkovska, J.; Krstanoski, L.; Veenman, L. Diagnostic and Therapeutic Potential of TSPO Studies Regarding Neurodegenerative Diseases, Psychiatric Disorders, Alcohol Use Disorders, Traumatic Brain Injury, and Stroke: An Update. Cells 2020, 9, 870. https://doi.org/10.3390/cells9040870
Dimitrova-Shumkovska J, Krstanoski L, Veenman L. Diagnostic and Therapeutic Potential of TSPO Studies Regarding Neurodegenerative Diseases, Psychiatric Disorders, Alcohol Use Disorders, Traumatic Brain Injury, and Stroke: An Update. Cells. 2020; 9(4):870. https://doi.org/10.3390/cells9040870
Chicago/Turabian StyleDimitrova-Shumkovska, Jasmina, Ljupcho Krstanoski, and Leo Veenman. 2020. "Diagnostic and Therapeutic Potential of TSPO Studies Regarding Neurodegenerative Diseases, Psychiatric Disorders, Alcohol Use Disorders, Traumatic Brain Injury, and Stroke: An Update" Cells 9, no. 4: 870. https://doi.org/10.3390/cells9040870